Spike-wave discharges are the hallmark of idiopathic generalized epilepsy. They are caused by a disorder in the thalamocortical network. Commercially available anti-epileptic drugs have pronounced side effects (i.e., sedation and gastroenterological concerns), which might result from a low selectivity to molecular targets. We suggest a specific subtype of adrenergic receptors (ARs) as a promising anti-epileptic molecular target. The influence of alpha2 ARs is mainly carried out through Gi/o-proteins although coupling to Gs was also demonstrated. Thus, activation of alpha2 ARs can either inhibit or stimulate different intracellular pathways. Numerous neuronal proteins interacting directly or indirectly with alpha2 ARs have been described. Here we describe some mechanisms of noradrenergic modulation of spike-wave activity via ion channels and integral membrane proteins.
- absence epilepsy
- spike-wave discharges
- adrenergic receptors
- alpha2 adrenergic receptors
1. Noradrenergic Regulation of Spike-Wave Activity
To date, a large amount of data has been accumulated on the noradrenergic modulation of spike-wave activity (e.g., [1][2][3][4]). Cortico-thalamo-cortical neural circuitry is known to receive dense innervations from noradrenergic neurons [5][6][7]. Nevertheless, existing anti-absence drugs do not directly affect noradrenaline-related mechanisms or molecular targets of noradrenaline (NA). Mechanisms of noradrenergic regulation of arousal level may play a significant role in the generation of SWDs. By participating in state transitions, the NA system sets favorable conditions at molecular, cellular, and network levels for the appearance of pathological spike-wave activity. We suppose that by separating a contribution of the noradrenergic system into sleep-wake cycling and its modulation of spike-wave activity, novel, more selective pharmacological targets could be identified.
In naturally falling asleep, the NA level gradually decreases [8] and the predominantly alpha1- and beta-AR-mediated excitatory effect of NA switches to an alpha2-mediated inhibitory effect. This state is favorable for triggering spike-wave activity [9][10] (the upper part in Figure 1) Natural awakening provides an almost instant turn to arousal with a very low probability of SWDs occurring [9][11]. Injections of alpha2 ARs agonists might cause an unnaturally rapid increase in activation of alpha2 ARs leading to an artificial predominance of alpha2-mediated effect over the effect of other ARs subtypes (the bottom part of Figure 1). The emergence from sedation is again an extended transition favorable for spike-wave discharges (SWDs) to occur. Future studies of pharmacologically induced absence status epilepticus are required to examine the dose-dependent effects of alpha2 ARs agonists on SWDs. Another intriguing issue regarding the role of each ARs subtypes in the modulation of spike-wave activity and arousal level.
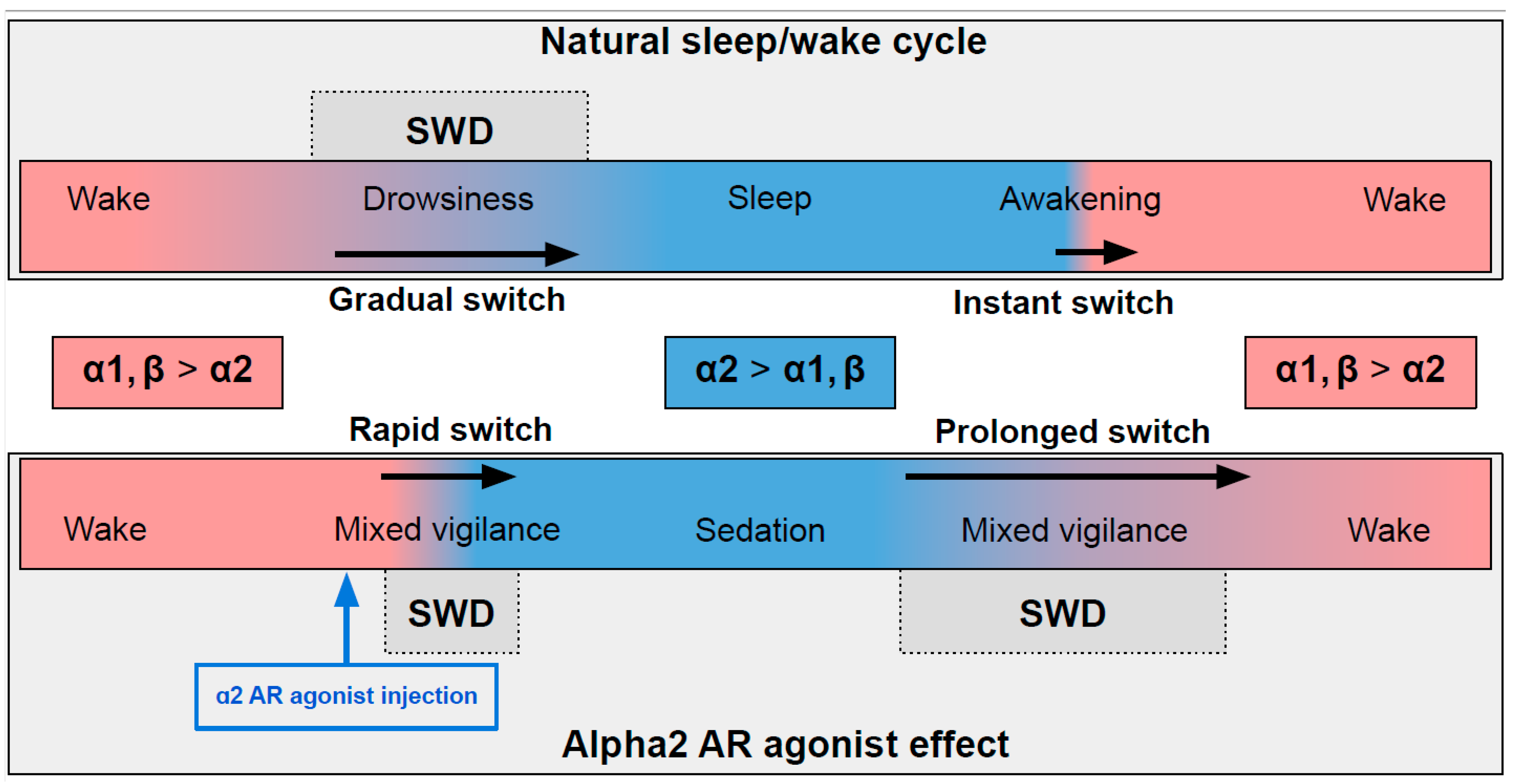
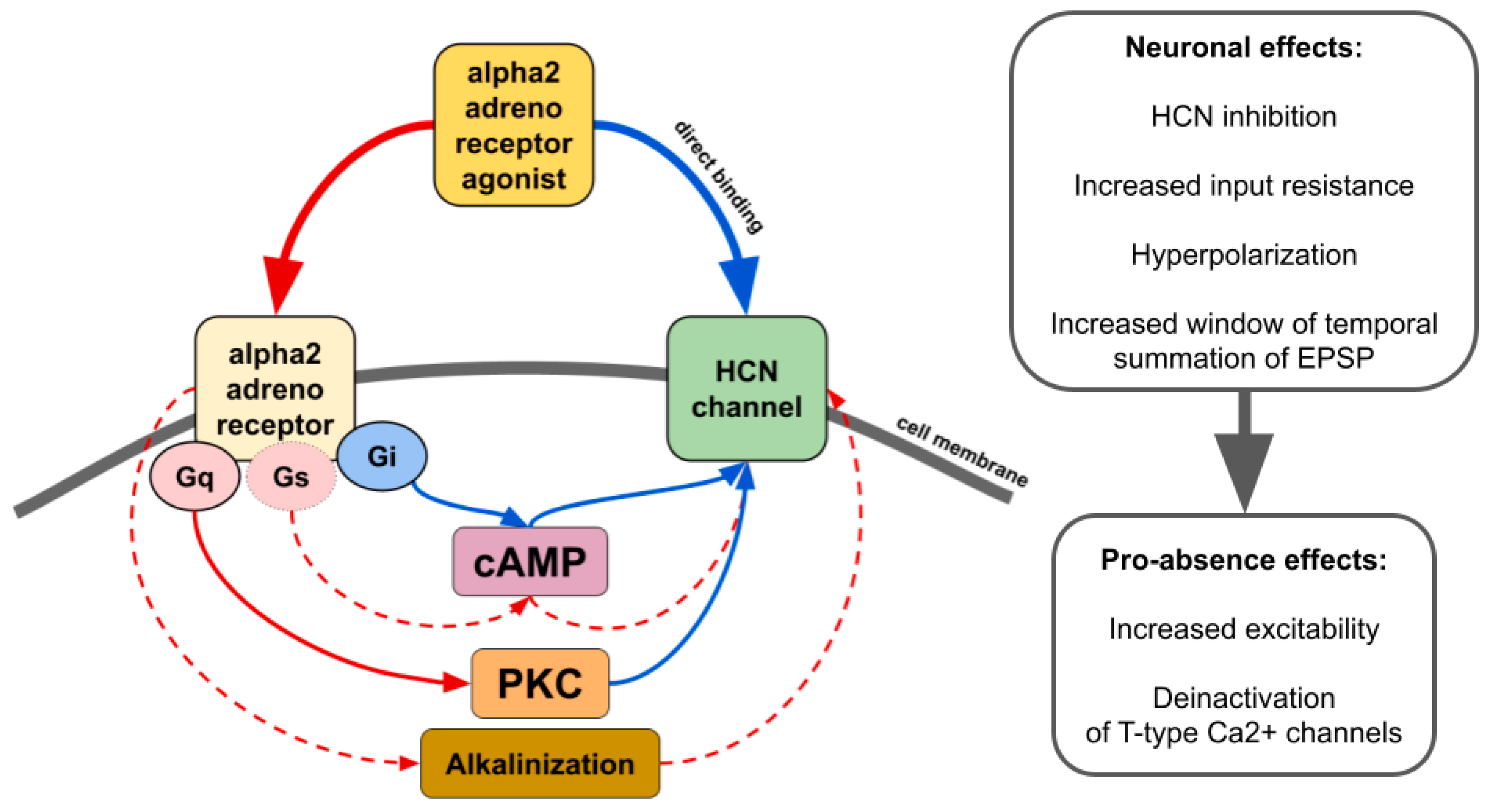
The influence of alpha2 ARs is mainly carried out through Gi/o-proteins although coupling to Gs was also demonstrated [12]. Thus, activation of alpha2 ARs can either inhibit or stimulate various intracellular pathways. Numerous neuronal proteins interacting directly or indirectly with alpha2 ARs have been described. Here we focus on phenomena related to spike-wave activity, such as interaction with ionic channels.
2. Alpha2-Adrenoreceptors and HCN Channels
3. Alpha2-Adrenoreceptors and Calcium Channels
4. Astrocytic Alpha2-Adrenoreceptors
This entry is adapted from the peer-reviewed paper 10.3390/ijms24021477
References
- Micheletti, G.; Warter, J.-M.; Marescaux, C.; Depaulis, A.; Tranchant, C.; Rumbach, L.; Vergnes, M. Effects of Drugs Affecting Noradrenergic Neurotransmission in Rats with Spontaneous Petit Mal-like Seizures. Eur. J. Pharmacol. 1987, 135, 397–402.
- Buzsáki, G.; Kennedy, B.; Solt, V.B.; Ziegler, M. Noradrenergic Control of Thalamic Oscillation: The Role of Alpha-2 Receptors. Eur. J. Neurosci. 1991, 3, 222–229.
- Sitnikova, E.; Luijtelaar, G. van Reduction of Adrenergic Neurotransmission with Clonidine Aggravates Spike-Wave Seizures and Alters Activity in the Cortex and the Thalamus in WAG/Rij Rats. Brain Res. Bull. 2005, 64, 533–540.
- Yavuz, M.; Aydın, B.; Çarçak, N.; Akman, Ö.; Raci Yananlı, H.; Onat, F. Atipamezole, a Specific A2A Antagonist, Suppresses Spike-and-Wave Discharges and Alters Ca2+ /Calmodulin-Dependent Protein Kinase II in the Thalamus of Genetic Absence Epilepsy Rats. Epilepsia 2020, 61, 2825–2835.
- Pérez-Santos, I.; Palomero-Gallagher, N.; Zilles, K.; Cavada, C. Distribution of the Noradrenaline Innervation and Adrenoceptors in the Macaque Monkey Thalamus. Cereb. Cortex 2021, 31, 4115–4139.
- Zhang, Y.; Fu, B.; Liu, C.; Yu, S.; Luo, T.; Zhang, L.; Zhou, W.; Yu, T. Activation of Noradrenergic Terminals in the Reticular Thalamus Delays Arousal from Propofol Anesthesia in Mice. FASEB J. 2019, 33, 7252–7260.
- Rho, H.-J.; Kim, J.-H.; Lee, S.-H. Function of Selective Neuromodulatory Projections in the Mammalian Cerebral Cortex: Comparison Between Cholinergic and Noradrenergic Systems. Front. Neural Circuits 2018, 12, 47.
- Kjaerby, C.; Andersen, M.; Hauglund, N.; Untiet, V.; Dall, C.; Sigurdsson, B.; Ding, F.; Feng, J.; Li, Y.; Weikop, P.; et al. Memory-enhancing properties of sleep depend on the oscillatory amplitude of norepinephrine. Nat Neurosci. 2022, 25, 1059–1070.
- Coenen, A.M.L.; Van Luijtelaar, E.L.J.M. Genetic Animal Models for Absence Epilepsy: A Review of the WAG/Rij Strain of Rats. Behav. Genet. 2003, 33, 635–655.
- Riekkinen, P.; Sirviö, J.; Jäkälä, P.; Lammintausta, R.; Riekkinen, P. Interaction between the Alpha 2-Noradrenergic and Muscarinic Systems in the Regulation of Neocortical High Voltage Spindles. Brain Res. Bull. 1990, 25, 147–149.
- Smyk, M.K.; van Luijtelaar, G. Circadian Rhythms and Epilepsy: A Suitable Case for Absence Epilepsy. Front. Neurol. 2020, 11, 245.
- Timmons, S.D.; Geisert, E.; Stewart, A.E.; Lorenzon, N.M.; Foehring, R.C. Alpha2-Adrenergic Receptor-Mediated Modulation of Calcium Current in Neocortical Pyramidal Neurons. Brain Res. 2004, 1014, 184–196.
- He, C.; Chen, F.; Li, B.; Hu, Z. Neurophysiology of HCN Channels: From Cellular Functions to Multiple Regulations. Prog. Neurobiol. 2014, 112, 1–23.
- Pape, H.C. Queer Current and Pacemaker: The Hyperpolarization-Activated Cation Current in Neurons. Annu. Rev. Physiol. 1996, 58, 299–327.
- Moosmang, S.; Biel, M.; Hofmann, F.; Ludwig, A. Differential Distribution of Four Hyperpolarization-Activated Cation Channels in Mouse Brain. Biol. Chem. 1999, 380, 975–980.
- Santoro, B.; Chen, S.; Luthi, A.; Pavlidis, P.; Shumyatsky, G.P.; Tibbs, G.R.; Siegelbaum, S.A. Molecular and Functional Heterogeneity of Hyperpolarization-Activated Pacemaker Channels in the Mouse CNS. J. Neurosci. 2000, 20, 5264–5275.
- Notomi, T.; Shigemoto, R. Immunohistochemical Localization of Ih Channel Subunits, HCN1-4, in the Rat Brain. J. Comp. Neurol. 2004, 471, 241–276.
- Wainger, B.J.; DeGennaro, M.; Santoro, B.; Siegelbaum, S.A.; Tibbs, G.R. Molecular Mechanism of CAMP Modulation of HCN Pacemaker Channels. Nature 2001, 411, 805–810.
- Chen, S.; Wang, J.; Siegelbaum, S.A. Properties of Hyperpolarization-Activated Pacemaker Current Defined by Coassembly of HCN1 and HCN2 Subunits and Basal Modulation by Cyclic Nucleotide. J. Gen. Physiol. 2001, 117, 491–504.
- Seifert, R.; Scholten, A.; Gauss, R.; Mincheva, A.; Lichter, P.; Kaupp, U.B. Molecular Characterization of a Slowly Gating Human Hyperpolarization-Activated Channel Predominantly Expressed in Thalamus, Heart, and Testis. Proc. Natl. Acad. Sci. USA 1999, 96, 9391–9396.
- Ishii, T.M.; Takano, M.; Xie, L.H.; Noma, A.; Ohmori, H. Molecular Characterization of the Hyperpolarization-Activated Cation Channel in Rabbit Heart Sinoatrial Node. J. Biol. Chem. 1999, 274, 12835–12839.
- Yavuz, M.; Onat, F. The Role of Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels in the Pathophysiology of Absence Epilepsy. J. Turkish Epilepsi Soc. 2018, 24, 41–50.
- Kuisle, M.; Wanaverbecq, N.; Brewster, A.L.; Frère, S.G.A.; Pinault, D.; Baram, T.Z.; Lüthi, A. Functional Stabilization of Weakened Thalamic Pacemaker Channel Regulation in Rat Absence Epilepsy. J. Physiol. 2006, 575, 83–100.
- Cain, S.M.; Tyson, J.R.; Jones, K.L.; Snutch, T.P. Thalamocortical Neurons Display Suppressed Burst-Firing Due to an Enhanced Ih Current in a Genetic Model of Absence Epilepsy. Pflügers Arch.–Eur. J. Physiol. 2015, 467, 1367–1382.
- David, F.; Çarçak, N.; Furdan, S.; Onat, F.; Gould, T.; Mészáros, Á.; Di Giovanni, G.; Hernández, V.M.; Chan, C.S.; Lőrincz, M.L.; et al. Suppression of Hyperpolarization-Activated Cyclic Nucleotide-Gated Channel Function in Thalamocortical Neurons Prevents Genetically Determined and Pharmacologically Induced Absence Seizures. J. Neurosci. 2018, 38, 6615–6627.
- Yavuz, M.; Aydın, B.; Çarçak, N.; Onat, F. Decreased Hyperpolarization-Activated Cyclic Nucleotide-Gated Channel 2 Activity in a Rat Model of Absence Epilepsy and the Effect of ZD7288, an Ih Inhibitor, on the Spike-and-Wave Discharges. Pharmacology 2022, 107, 227–234.
- Strauss, U.; Kole, M.H.P.; Bräuer, A.U.; Pahnke, J.; Bajorat, R.; Rolfs, A.; Nitsch, R.; Deisz, R.A. An Impaired Neocortical Ih Is Associated with Enhanced Excitability and Absence Epilepsy. Eur. J. Neurosci. 2004, 19, 3048–3058.
- Kole, M.H.P.; Bräuer, A.U.; Stuart, G.J. Inherited Cortical HCN1 Channel Loss Amplifies Dendritic Calcium Electrogenesis and Burst Firing in a Rat Absence Epilepsy Model. J. Physiol. 2007, 578, 507–525.
- Zobeiri, M.; Chaudhary, R.; Blaich, A.; Rottmann, M.; Herrmann, S.; Meuth, P.; Bista, P.; Kanyshkova, T.; Lüttjohann, A.; Narayanan, V.; et al. The Hyperpolarization-Activated HCN4 Channel Is Important for Proper Maintenance of Oscillatory Activity in the Thalamocortical System. Cereb. Cortex 2019, 29, 2291–2304.
- Lörincz, A.; Notomi, T.; Tamás, G.; Shigemoto, R.; Nusser, Z. Polarized and Compartment-Dependent Distribution of HCN1 in Pyramidal Cell Dendrites. Nat. Neurosci. 2002, 5, 1185–1193.
- Huang, Z.; Walker, M.C.; Shah, M.M. Loss of Dendritic HCN1 Subunits Enhances Cortical Excitability and Epileptogenesis. J. Neurosci. 2009, 29, 10979–10988.
- Blumenfeld, H.; Klein, J.P.; Schridde, U.; Vestal, M.; Rice, T.; Khera, D.S.; Bashyal, C.; Giblin, K.; Paul-Laughinghouse, C.; Wang, F.; et al. Early Treatment Suppresses the Development of Spike-Wave Epilepsy in a Rat Model. Epilepsia 2008, 49, 400–409.
- Yang, Y.; Meng, Q.; Pan, X.; Xia, Z.; Chen, X. Dexmedetomidine Produced Analgesic Effect via Inhibition of HCN Currents. Eur. J. Pharmacol. 2014, 740, 560–564.
- Carr, D.B.; Andrews, G.D.; Glen, W.B.; Lavin, A. Alpha2-Noradrenergic Receptors Activation Enhances Excitability and Synaptic Integration in Rat Prefrontal Cortex Pyramidal Neurons via Inhibition of HCN Currents. J. Physiol. 2007, 584, 437–450.
- Näsman, J.; Jansson, C.C.; Akerman, K.E. The Second Intracellular Loop of the Alpha2-Adrenergic Receptors Determines Subtype-Specific Coupling to CAMP Production. J. Biol. Chem. 1997, 272, 9703–9708.
- Won, J.; Lee, P.R.; Oh, S.B. Alpha 2 Adrenoceptor Agonist Guanabenz Directly Inhibits Hyperpolarization-Activated, Cyclic Nucleotide-Modulated (HCN) Channels in Mesencephalic Trigeminal Nucleus Neurons. Eur. J. Pharmacol. 2019, 854, 320–327.
- Wang, M.; Ramos, B.P.; Paspalas, C.D.; Shu, Y.; Simen, A.; Duque, A.; Vijayraghavan, S.; Brennan, A.; Dudley, A.; Nou, E.; et al. Alpha2A-Adrenoceptors Strengthen Working Memory Networks by Inhibiting CAMP-HCN Channel Signaling in Prefrontal Cortex. Cell 2007, 129, 397–410.
- Parkis, M.A.; Berger, A.J. Clonidine Reduces Hyperpolarization-Activated Inward Current (Ih) in Rat Hypoglossal Motoneurons. Brain Res. 1997, 769, 108–118.
- Brummett, C.M.; Hong, E.K.; Janda, A.M.; Amodeo, F.S.; Lydic, R. Perineural Dexmedetomidine Added to Ropivacaine for Sciatic Nerve Block in Rats Prolongs the Duration of Analgesia by Blocking the Hyperpolarization-Activated Cation Current. Anesthesiology 2011, 115, 836–843.
- Ramos, B.P.; Stark, D.; Verduzco, L.; van Dyck, C.H.; Arnsten, A.F.T. A2A-Adrenoceptor Stimulation Improves Prefrontal Cortical Regulation of Behavior through Inhibition of CAMP Signaling in Aging Animals. Learn. Mem. 2006, 13, 770–776.
- Isom, L.L.; Cragoe, E.J.; Limbird, L.E. Alpha 2-Adrenergic Receptors Accelerate Na+/H+ Exchange in Neuroblastoma X Glioma Cells. J. Biol. Chem. 1987, 262, 6750–6757.
- Munsch, T.; Pape, H.C. Modulation of the Hyperpolarization-Activated Cation Current of Rat Thalamic Relay Neurones by Intracellular PH. J. Physiol. 1999, 519 Pt 2, 493–504.
- Salvati, K.A.; Souza, G.M.P.R.; Lu, A.C.; Ritger, M.L.; Guyenet, P.; Abbott, S.B.; Beenhakker, M.P. Respiratory Alkalosis Provokes Spike-Wave Discharges in Seizure-Prone Rats. Elife 2022, 11, e72898.
- Lüscher, C.; Slesinger, P.A. Emerging Roles for G Protein-Gated Inwardly Rectifying Potassium (GIRK) Channels in Health and Disease. Nat. Rev. Neurosci. 2010, 11, 301–315.
- Shirasaka, T.; Kannan, H.; Takasaki, M. Activation of a G Protein–Coupled Inwardly Rectifying K+Current and Suppression of I HContribute to Dexmedetomidine-Induced Inhibition of Rat Hypothalamic Paraventricular Nucleus Neurons. Anesthesiology 2007, 107, 605–615.
- Kobayashi, T.; Hirai, H.; Iino, M.; Fuse, I.; Mitsumura, K.; Washiyama, K.; Kasai, S.; Ikeda, K. Inhibitory Effects of the Antiepileptic Drug Ethosuximide on G Protein-Activated Inwardly Rectifying K+ Channels. Neuropharmacology 2009, 56, 499–506.
- Simms, B.A.; Zamponi, G.W. Neuronal Voltage-Gated Calcium Channels: Structure, Function, and Dysfunction. Neuron 2014, 82, 24–45.
- Van Luijtelaar, G.; Wiaderna, D.; Elants, C.; Scheenen, W. Opposite Effects of T- and L-Type Ca2+ Channels Blockers in Generalized Absence Epilepsy. Eur. J. Pharmacol. 2000, 406, 381–389.
- Sadighi, M.; Shahabi, P.; Gorji, A.; Pakdel, F.G.; Nejad, G.G.; Ghorbanzade, A. Role of L- and T-Type Calcium Channels in Regulation of Absence Seizures in Wag/Rij Rats. Neurophysiology 2013, 45, 312–318.
- Nacif-Coelho, C.; Correa-Sales, C.; Chang, L.L.; Maze, M. Perturbation of Ion Channel Conductance Alters the Hypnotic Response to the A2-Adrenergic Agonist Dexmedetomidine in the Locus Coeruleus of the Rat. Anesthesiology 1994, 81, 1527–1534.
- Desai, M.K.; Dikshit, R.K.; Mansuri, S.M.; Shah, U.H. Effect of Nifedipine, a Calcium Channel Inhibitor, on Sedation Produced by Reserpine, Clonidine and Propranolol in Mice. Indian J. Exp. Biol. 1994, 32, 314–317.
- Reid, K.; Guo, T.Z.; Davies, M.F.; Maze, M. Nifedipine, an L-Type Calcium Channel Blocker, Restores the Hypnotic Response in Rats Made Tolerant to the Alpha-2 Adrenergic Agonist Dexmedetomidine. J. Pharmacol. Exp. Ther. 1997, 283, 993–999.
- Czarnecka, E.; Tymczyszyn, W.; Pietrzak, B. Effect of Nifedipine and Verapamil on Hypotensive Action of Clonidine in Rabbits. Pol. J. Pharmacol. 1998, 50, 193–201.
- Nehlig, A.; Vergnes, M.; Waydelich, R.; Hirsch, E.; Charbonne, R.; Marescaux, C.; Seylaz, J. Absence Seizures Induce a Decrease in Cerebral Blood Flow: Human and Animal Data. J. Cereb. Blood Flow Metab. 1996, 16, 147–155.
- Kolaj, M.; Renaud, L. Norepinephrine Acts via A2 Adrenergic Receptors to Suppress N-Type Calcium Channels in Dissociated Rat Median Preoptic Nucleus Neurons. Neuropharmacology 2001, 41, 472–480.
- Hara, M.; Zhou, Z.Y.; Hemmings, H.C. A2-Adrenergic Receptor and Isoflurane Modulation of Presynaptic Ca2+ Influx and Exocytosis in Hippocampal Neurons. Anesthesiology 2016, 125, 535–546.
- Tokuda, S.; Kuramoto, T.; Tanaka, K.; Kaneko, S.; Takeuchi, I.K.; Sasa, M.; Serikawa, T. The Ataxic Groggy Rat Has a Missense Mutation in the P/Q-Type Voltage-Gated Ca2+ Channel A1A Subunit Gene and Exhibits Absence Seizures. Brain Res. 2007, 1133, 168–177.
- Bomben, V.C.; Aiba, I.; Qian, J.; Mark, M.D.; Herlitze, S.; Noebels, J.L. Isolated P/Q Calcium Channel Deletion in Layer VI Corticothalamic Neurons Generates Absence Epilepsy. J. Neurosci. 2016, 36, 405–418.
- McCormick, D.A.; Connors, B.W.; Lighthall, J.W.; Prince, D.A. Comparative Electrophysiology of Pyramidal and Sparsely Spiny Stellate Neurons of the Neocortex. J. Neurophysiol. 1985, 54, 782–806.
- Llinás, R.; Jahnsen, H. Electrophysiology of Mammalian Thalamic Neurones in Vitro. Nature 1982, 297, 406–408.
- Lee, S.E.; Lee, J.; Latchoumane, C.; Lee, B.; Oh, S.-J.; Saud, Z.A.; Park, C.; Sun, N.; Cheong, E.; Chen, C.-C.; et al. Rebound Burst Firing in the Reticular Thalamus Is Not Essential for Pharmacological Absence Seizures in Mice. Proc. Natl. Acad. Sci. USA 2014, 111, 11828–11833.
- McCafferty, C.; David, F.; Venzi, M.; Lőrincz, M.L.; Delicata, F.; Atherton, Z.; Recchia, G.; Orban, G.; Lambert, R.C.; Di Giovanni, G.; et al. Cortical Drive and Thalamic Feed-Forward Inhibition Control Thalamic Output Synchrony during Absence Seizures. Nat. Neurosci. 2018, 21, 744–756.
- Coulter, D.A.; Steinhauser, C. Role of Astrocytes in Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022434.
- Verhoog, Q.P.; Holtman, L.; Aronica, E.; van Vliet, E.A. Astrocytes as Guardians of Neuronal Excitability: Mechanisms Underlying Epileptogenesis. Front. Neurol. 2020, 11, 1541.
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and Epilepsy: Excitability and Inflammation. Trends Neurosci. 2013, 36, 174–184.
- Binder, D.K.; Steinhäuser, C. Functional Changes in Astroglial Cells in Epilepsy. Glia 2006, 54, 358–368.
- Ozgur, M.; Özyurt, M.G.; Arkan, S.; Cavdar, S. The Effects of Optogenetic Activation of Astrocytes on Spike-and-Wave Discharges in Genetic Absence Epileptic Rats. Ann. Neurosci. 2022, 29, 53–61.
- Hertz, L.; Lovatt, D.; Goldman, S.A.; Nedergaard, M. Adrenoceptors in Brain: Cellular Gene Expression and Effects on Astrocytic Metabolism and I. Neurochem. Int. 2010, 57, 411–420.
- Gaidin, S.G.; Zinchenko, V.P.; Sergeev, A.I.; Teplov, I.Y.; Mal’tseva, V.N.; Kosenkov, A.M. Activation of Alpha-2 Adrenergic Receptors Stimulates GABA Release by Astrocytes. Glia 2020, 68, 1114–1130.
- Liu, Z.; Vergnes, M.; Depaulis, A.; Marescaux, C. Evidence for a Critical Role of GABAergic Transmission within the Thalamus in the Genesis and Control of Absence Seizures in the Rat. Brain Res. 1991, 545, 1–7.
- Liu, Z.; Vergnes, M.; Depaulis, A.; Marescaux, C. Involvement of Intrathalamic GABAb Neurotransmission in the Control of Absence Seizures in the Rat. Neuroscience 1992, 48, 87–93.
- Vitellaro-Zuccarello, L.; Calvaresi, N.; De Biasi, S. Expression of GABA Transporters, GAT-1 and GAT-3, in the Cerebral Cortex and Thalamus of the Rat during Postnatal Development. Cell Tissue Res. 2003, 313, 245–257.