G protein-coupled receptors (GPCRs) are cell surface receptors that mediate the function of extracellular ligands. Understanding how GPCRs work at the molecular level has important therapeutic implications, as 30-40% of the drugs currently in clinical use mediate therapeutic effects by acting on GPCRs. Like many other cell types, hepatocyte's function is regulated by GPCRs. A better understanding of the metabolic role of GPCRs in hepatocytes, the dominant constituent cells of the liver, could lead to the development of novel drugs that are clinically useful for the treatment of various metabolic diseases, including Type 2 diabetes, NAFLD, and NASH.
- GPCR, Hepatocyte, Liver, NAFLD, Diabetes
1. Introduction
The liver is the crucial orchestrator of metabolism. In humans, approximately 11,000 genes are actively transcribed in hepatocytes, and many of them are related to the metabolic functions of the liver [1]. The liver mediates numerous physiological functions, including protein synthesis, macronutrient metabolism, cholesterol and lipid homeostasis, endocrine control of growth signaling pathways, and drug detoxification [2][3][4][5][6]. Metabolic hormones, nutrients, and sympathetic and parasympathetic neural signals tightly regulate liver function [7][8]. Alterations in these signaling pathways lead to the dysfunction of liver metabolism and can cause insulin resistance, type 2 diabetes (T2D), nonalcoholic fatty liver disease (NAFLD), nonalcoholic steatohepatitis (NASH), and cancer [7][8][9]. The liver consists of hepatocytes, biliary epithelial cells, stellate cells, smooth muscle cells, vascular endothelial cells, various immune cells, and sinusoidal endothelial cells [10][11][12]. Each of these cell types possesses a unique function and collectively regulates liver function at multiple levels. Hepatocytes are the primary cell type present in the liver and account for 80% of the liver cell population [12].
G protein-coupled receptors (GPCRs) are cell surface receptors and mediate the function of a vast number of extracellular ligands (neurotransmitters, hormones, etc.) [13,14]. Ligands-coupled GPCRs recognize and activate heterotrimeric G proteins, which comprise Gα, Gβ, and Gγ. Gα determines the functions of a G protein through the exchange of guanosine diphosphate (G[13][14]DP) with guanosine triphosphate (GTP) [15]. The Gβ and Gγ subunits are separately synthesized and form a complex to function as a single functional unit [16]. Both the Gα and Gβγ subunits activate signaling molecules and trigger cellular responses. Based on their α subunit, G proteins are classified into four families: Gs, Gi, Gq, and G12/13 (Figure 1), where s and i represent stimulation and inhibition, respectively. Receptor-activated G proteins stimulate various downstream signaling pathways; for example, Gs and Gi proteins control adenylyl cyclase activity, Gq activates phospholipase Cβ, and G12/13 stimulates guanine nucleotide exchange factors for small GTPases of the Rho family [17][18]. The human genome contains about 800 different GPCR genes, which represent 3–4% of all human genes. Importantly, 30–40% of the drugs currently used in the clinic mediate their therapeutic effects via acting on GPCRs [13]. Understanding how GPCRs work at the molecular level is, therefore, of considerable therapeutic relevance.
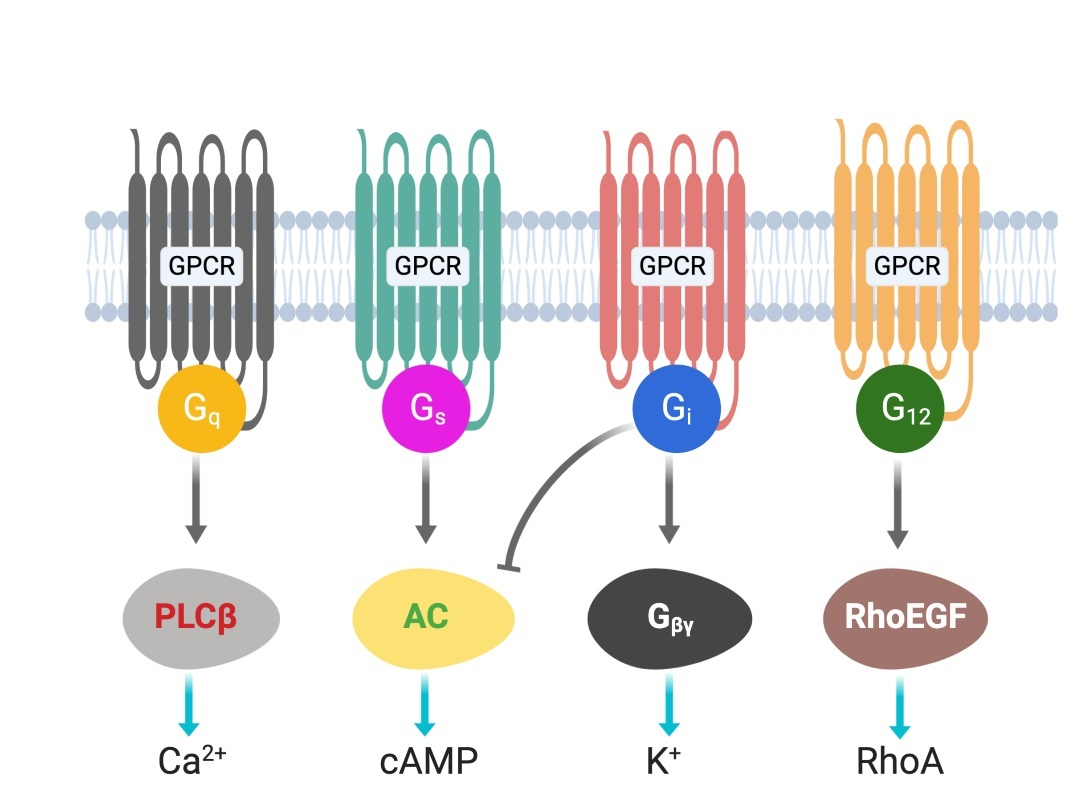
Figure 1. G-proteins’ classification and downstream signals.
2. Role of GPCRs in Liver Metabolism
2.1. Gs-Coupled GPCRs
The glucagon receptor (GCGR) is abundantly expressed by hepatocytes and is the most extensively studied Gs-coupled GPCR in the liver [19]. GCGR is activated by the 29 amino acid polypeptide glucagon, which is secreted by β cells of the endocrine pancreas, in particular during the fasted state or exercise [20]. The primary function of glucagon is to prevent hypoglycemia during fasting by increasing hepatic glucose production (HGP) from hepatocytes by stimulating both glycogenolysis and gluconeogenesis [21][22]. Activation of GCGR in hepatocytes results in increased intracellular cyclic adenosine monophosphate (cAMP) levels, followed by activation of protein kinase A (PKA) and cAMP response element-binding (CREB) protein (Figure 2). Phosphorylated CREB (the active form of CREB) binds to CRE and increases the expression of essential gluconeogenic genes, including glucose 6-phosphatase (G6pc) and phosphoenolpyruvate carboxykinase (Pck1) [23]. The activated GCGR also stimulates Ca2+ release from the endoplasmic reticulum (ER) and activates calcium/calmodulin-dependent protein kinase II (CAMKII). This results in the translocation of FOXO1 to the nucleus that promotes the transcription of gluconeogenic genes[24]. In addition to transcriptional regulation of genes involved in glucose metabolism, GCGR activation also modulates the activity of enzymes involved in glucose metabolism via PKA-dependent phosphorylation. PKA phosphorylates phospho-fructokinase 2 (PFK-2), fructose 2,6-bisphosphatase (FBPase2), pyruvate kinase, and phosphorylase kinase. Phosphorylation of these enzymes increases HGP by increasing gluconeogenesis and glycogenolysis or by inhibiting glycolysis [25].
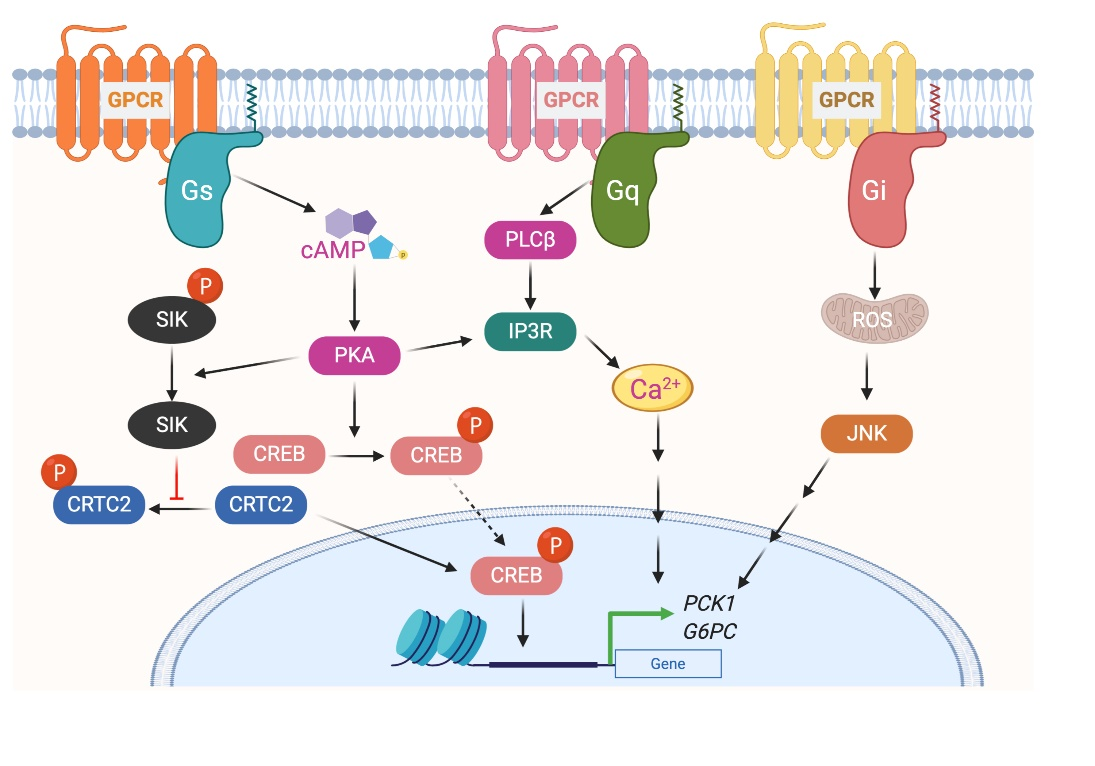
Figure 2. G protein-coupled receptors-mediated glycogenesis in hepatocytes.
2.2. Gi-Coupled GPCRs
GPCRs that preferentially couple to Gi proteins inhibit the function of adenylyl cyclase and thereby decrease intracellular cAMP. In a recent study, to understand the role of hepatic Gi signaling on whole-body glucose metabolism, Dr. Wess's group employed a chemo-genetic approach where a designer Gi-coupled receptor was selectively expressed in mouse hepatocytes (Hep-GiD). This designer receptor can be selectively activated by clozapine-N-oxide (CNO), an otherwise chemically inert compound [26]. As activation of the Gs-coupled GCGR increases HGP, it was expected that activation of Gi in the liver would have opposite effects. Interestingly, activation of Gi signaling in hepatocytes also increased HGP by activating c-Jun N-terminal kinases (JNK) signaling [26]. Activation of Gi signaling in mouse liver impaired glucose tolerance and stimulated gluconeogenesis and glycogenolysis. Both in vivo and in vitro studies showed that enhanced Gi signaling in hepatocytes led to a JNK-dependent increase in the expression of G6pc (Figure 2). Similar results were obtained with the human hepatocytes. Activation of Gi signaling in the liver did not affect hepatic insulin signaling [26].
2.3. Gq-Coupled GPCRs
Stimulation of Gq signaling leads to the activation of phospholipase Cβ, followed by an increase in intracellular levels of inositol trisphosphate, diacylglycerol, and calcium (Figure 1). Further downstream effectors of the signaling pathway are dependent on the receptor and cell type. To understand the role of hepatic Gq signaling on glucose homeostasis, Dr. Wess's group expressed a Gq coupled, CNO-sensitive designer receptor selectively in mouse hepatocytes (Hep-GqD)[27]. Acute activation of this receptor with CNO increased blood glucose levels in a concentration-dependent manner. Hep-GqD mice showed impaired glucose and pyruvate tolerance. Impairments were predominately due to an increase in HGP [26]. Mechanistic studies confirmed an increase in the expression of various genes involved in gluconeogenesis and glycogenolysis. In particular, mRNA levels of Pck1, a rate-limiting enzyme of gluconeogenesis, were significantly increased (Figure 2). Similarly, the expression levels of genes involved in fatty acid syntheses such as Acyl, Acc, and Fas were also upregulated [26]. These data indicate that Gq signaling in hepatocytes plays a crucial role in glucose and lipid handling.
*The references listed here are based on the original article.
This entry is adapted from the peer-reviewed paper 10.3390/biom10101445
References
- Ramskold, D.; Wang, E.T.; Burge, C.B.; Sandberg, R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 2009, 5, e1000598. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Hers, H.G. The control of glycogen metabolism in the liver. Annu. Rev. Biochem. 1976, 45, 167–189. [Google Scholar] [CrossRef]
- Rines, A.K.; Sharabi, K.; Tavares, C.D.; Puigserver, P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat. Rev. Drug Discov. 2016, 15, 786–804. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Dentin, R.; Girard, J. Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab. 2004, 30, 398–408. [Google Scholar] [CrossRef]
- Grant, D.M. Detoxification pathways in the liver. J. Inherit. Metab. Dis. 1991, 14, 421–430. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Gordillo, M.; Evans, T.; Gouon-Evans, V. Orchestrating liver development. Development 2015, 142, 2094–2108. [Google Scholar] [CrossRef]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Ding, C.; Li, Y.; Guo, F.; Jiang, Y.; Ying, W.; Li, D.; Yang, D.; Xia, X.; Liu, W.; Zhao, Y.; et al. A Cell-type-resolved Liver Proteome. Mol. Cell Proteom. 2016, 15, 3190–3202. [Google Scholar] [CrossRef] [PubMed]
- Kmiec, Z. Cooperation of liver cells in health and disease. Adv. Anat. Embryol. Cell Biol. 2001, 161, III–XIII, 1–151. [Google Scholar] [CrossRef]
- Lagerstrom, M.C.; Schioth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 2004, 25, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Denis, C.; Sauliere, A.; Galandrin, S.; Senard, J.M.; Gales, C. Probing heterotrimeric G protein activation: Applications to biased ligands. Curr. Pharm. Des. 2012, 18, 128–144. [Google Scholar] [CrossRef]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, J.J. The quest to understand heterotrimeric G protein signaling. Nat. Struct. Mol. Biol. 2010, 17, 650–652. [Google Scholar] [CrossRef]
- Jastrzebska, B. GPCR: G protein complexes--the fundamental signaling assembly. Amino Acids 2013, 45, 1303–1314. [Google Scholar] [CrossRef]
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sorensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Glucagon Resistance. Int. J. Mol. Sci. 2019, 20, 3314. [Google Scholar] [CrossRef] [PubMed]
- Wewer Albrechtsen, N.J.; Pedersen, J.; Galsgaard, K.D.; Winther-Sorensen, M.; Suppli, M.P.; Janah, L.; Gromada, J.; Vilstrup, H.; Knop, F.K.; Holst, J.J. The Liver-alpha-Cell Axis and Type 2 Diabetes. Endocr. Rev. 2019, 40, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Ramnanan, C.J.; Edgerton, D.S.; Kraft, G.; Cherrington, A.D. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes. Metab. 2011, 13 (Suppl. 1), 118–125. [Google Scholar] [CrossRef] [PubMed]
- Felig, P.; Wahren, J.; Sherwin, R.; Hendler, R. Insulin, glucagon, and somatostatin in normal physiology and diabetes mellitus. Diabetes 1976, 25, 1091–1099. [Google Scholar] [CrossRef]
- Berglund, E.D.; Lee-Young, R.S.; Lustig, D.G.; Lynes, S.E.; Donahue, E.P.; Camacho, R.C.; Meredith, M.E.; Magnuson, M.A.; Charron, M.J.; Wasserman, D.H. Hepatic energy state is regulated by glucagon receptor signaling in mice. J. Clin. Investig. 2009, 119, 2412–2422. [Google Scholar] [CrossRef]
- Ozcan, L.; Wong, C.C.; Li, G.; Xu, T.; Pajvani, U.; Park, S.K.; Wronska, A.; Chen, B.X.; Marks, A.R.; Fukamizu, A.; et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012, 15, 739–751. [Google Scholar] [CrossRef]
- Galsgaard, K.D.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Lipid Metabolism. Front. Physiol. 2019, 10, 413. [Google Scholar] [CrossRef]
- Rossi, M.; Zhu, L.; McMillin, S.M.; Pydi, S.P.; Jain, S.; Wang, L.; Cui, Y.; Lee, R.J.; Cohen, A.H.; Kaneto, H.; et al. Hepatic Gi signaling regulates whole-body glucose homeostasis. J. Clin. Investig. 2018, 128, 746–759. [Google Scholar] [CrossRef]
- Li, J.H.; Jain, S.; McMillin, S.M.; Cui, Y.; Gautam, D.; Sakamoto, W.; Lu, H.; Jou, W.; McGuinness, O.P.; Gavrilova, O.; et al. A novel experimental strategy to assess the metabolic effects of selective activation of a G(q)-coupled receptor in hepatocytes in vivo. Endocrinology 2013, 154, 3539–3551. [Google Scholar] [CrossRef]
- Li, J.H.; Jain, S.; McMillin, S.M.; Cui, Y.; Gautam, D.; Sakamoto, W.; Lu, H.; Jou, W.; McGuinness, O.P.; Gavrilova, O.; et al. A novel experimental strategy to assess the metabolic effects of selective activation of a G(q)-coupled receptor in hepatocytes in vivo. Endocrinology 2013, 154, 3539–3551. [Google Scholar] [CrossRef]