1. Protocols for Colon Organoid Culture
1.1. Establishing Normal and Tumour Organoids from Colon Tissues
In 2011, Sato et al. adapted a protocol for growing murine intestinal organoids to successfully culture human normal colon, Barrett’s epithelium, and colon adenoma and adenocarcinoma organoids
[1]. Normal colon organoids require a wide range of media supplements to mimic the influence of signalling and niche factors, including Wnt3A, R-spondin-1/3 and Noggin
[1]. However, colon tumour organoids possess mutations that render certain factors unnecessary. For example, the adenomatous polyposis coli (
APC) gene is the most mutated gene in colon cancer and leads to constitutive Wnt signalling; this removes the dependence of the tumour on Wnt agonists (Wnt3A and R-spondins).
The L-WRN cell line (ATCC CRL-3276) was created by stably transfecting the Wnt3A-producing L-Wnt3A cell line (ATCC CRL-2647) with a vector expressing R-spondin-3 and Noggin
[2]. Conditioned media harvested from this cell line contains these three factors with greater stability and at lower cost than if they were reconstituted from a lyophilised powder
[2]. Batches of conditioned medium from L-WRN cells are highly reproducible
[3].
Our protocol for generating organoids from patient-derived colon tumour tissue and patient-matched normal colonic mucosa will be outlined. This is based on methods developed and optimised previously by laboratories worldwide
[1][4][5][6][7][8].
Tissue specimens are received from local hospitals on ice on the day of surgery, and a sample is taken for histological analysis. Colon tumour tissue is washed with PBS, minced into a paste using dissecting scissors and incubated in 10 mL of digestion buffer (1 mg/mL collagenase VI and 0.2 mg/mL DNase I) at 37 °C for 45–60 min. The digest is mechanically disrupted every 15 min using a 10 mL serological pipette by pipetting up and down ~10 times, expelling the contents with the tip almost flat on the bottom of the dish, until cells have been sufficiently released. The reaction is stopped by adding 1 mL of foetal calf serum and 10 mL of PBS, the digest is filtered through a 100 µM filter to remove large chunks, connective tissue, and mucus, and a red blood cell lysis is performed using a buffer of ammonium chloride (155 mM), sodium bicarbonate (12 mM), and EDTA (0.1 mM). Cells are pelleted by centrifugation at 400 g and 4 °C for 5 min, counted, and embedded in 40 µL domes of Cultrex® 3D RGF BME Culture Matrix (R&D Systems, #3445-005-01), a basement membrane gel composed primarily of laminin and collagen IV and optimised for the development of 3D organoid structures, at a density of ~1000 cells per µL (~40,000 cells per dome). In each well of a standard 24-well culture plate, 1–2 domes are seeded.
When processing samples of normal colon, the mucosa is isolated from the investing muscle layer using dissecting scissors, cut into smaller pieces (~2–5 mm) and washed with PBS by pipetting up and down ~10 times. Tissue pieces are incubated in EDTA buffer (7.5 mM EDTA in PBS with antibiotic-antimycotic [penicillin, streptomycin, amphotericin B]) at 4°C on a rocker for 60 min. Wash steps are then performed using 10 mL of PBS and a 10 mL serological pipette, by drawing the tissue pieces up and down 10 times to release crypts, retaining the 10 mL of PBS wash buffer in a 50 mL tube lined with 1% BSA, and then repeating this wash procedure a further 5 times to give a total of 50 mL of PBS containing isolated crypts. The crypts are pelleted by centrifugation at 200 g and 4 °C for 3 min, counted, and seeded in Cultrex domes at a density of 50–200 crypts per dome.
Organoids generated from the stem cells of normal colon tissue form cystic structures consisting of a hollow lumen surrounded by columnar epithelium and can exhibit a budding pattern (
Figure 1)
[1]. Noggin, a BMP inhibitor, is required for the maintenance of LGR5+ colon stem cells within the organoids
[1]. Nicotinamide is essential for extending organoid growth beyond 1 week
[1]. Furthermore, A83-01 (TGF-β receptor [Alk4/5/7] inhibitor) and either SB202190 (p38 inhibitor) or IGF1 + FGF2 allow the organoids to persist for at least 6 months
[1]. Removing Wnt3A, R-spondin, Noggin, and Nicotinamide from the culture medium, and adding the γ-secretase inhibitor DAPT, causes organoids to differentiate
[1][8].
Figure 1. Representative images of normal colon organoids: (A) on day 7 of passage 1 showing cystic organoids with large hollow lumens; (B) on day 8 of passage 1 showing cystic organoids with large hollow lumens; (C) on day 7 of passage 2 showing more compact organoids; (D) on day 5 of passage 6 showing epithelial budding. Original magnification: 4× (A–C) and 10× (D).
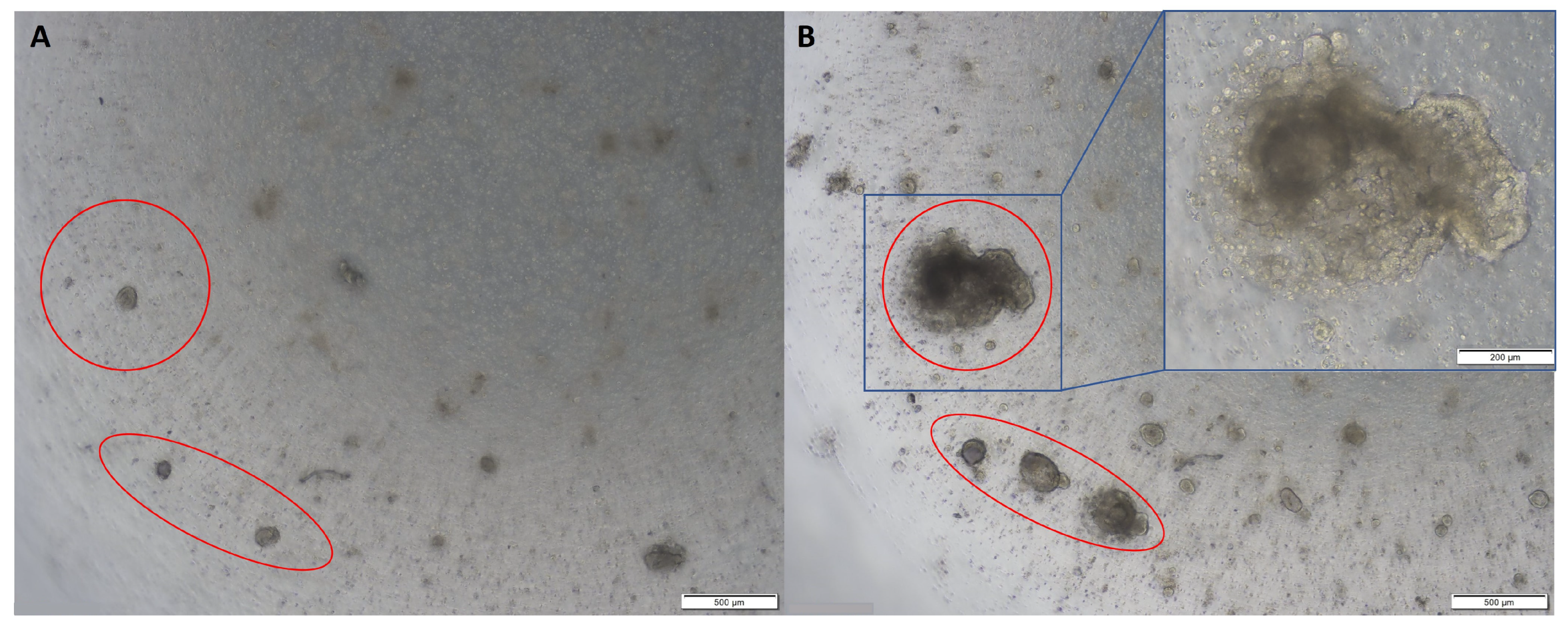
Colon cancer organoids tend to develop as compact masses of cells without the lumens observed in normal colon organoids (
Figure 2)
[1]. Most colon tumour organoids do not require Noggin in the culture medium
[1]. Approximately 80% of colon cancers have mutations to
APC,
AXIN2,
CTNNB1, or
TCF7L2 causing constitutive Wnt signalling, thus removing the requirement for Wnt3A or R-spondin; the remaining 20% have wild-type
APC genes and are, therefore, dependent on the addition of Wnt3A in the culture medium
[9]. Furthermore, some colon tumour organoids require both exogenous EGF and SB202190, and others require neither
[9]. Fujii et al. suggest trialling each new organoid culture on an array of different media formulations to determine the optimal conditions for each individual sample
[9].
Figure 2. Representative images of colon tumour organoids: (A) on day 2 of passage 2; (B) on day 10 of passage 2. Original magnification: 4× and 10× (inset).
Characterisation is a vital aspect of organoid development and validation. To validate stem cell-derived organoids and ensure that they contain a heterogeneous population of cells, markers of the various colon cell types can be visualised using fluorescence confocal microscopy
[10][11]. Colon stem cells give rise to the other colonic cell types and express LGR5 on their surface
[12]. They are supported by Paneth cells that express LYZ and give rise to transit amplifying cells that express EPHB2. The transit amplifying cells differentiate into the other colonic cell types as they migrate up from the crypt base towards the lumenal epithelial surface. Goblet cells are secretory cells, identified by their expression of MUC2. The absorptive cells, enterocytes, can be visualised by their expression of Villin (VIL1) or CA2. CHGA is used to identify colonic endocrine cells. The epithelial barrier function of organoids can be assayed using an uptake assay using FITC-labelled dextran
[6].
1.3. Reversed-Polarity Organoids
The apical surface of the epithelium that faces the lumen of the gut is on the interior surface of the organoid. This limits the validity of organoid assays, such as drug exposure and bacterial co-culture, because the pertinent interface is not accessible if factors are simply added to the media. The apical surface of epithelial cells facilitates absorption, secretion, and binding/detection, whereas the basolateral surface is responsible for anchorage, nutrient delivery to the bloodstream, and intercellular communication
[6]. This issue has been circumvented by microinjection of drugs or bacteria into the organoid lumen, or alternatively by breaking up the organoids, adding the drug or bacteria to the culture medium, and allowing the organoids to reform
[13][14]. However, microinjection requires a high degree of technical expertise, and breaking and reforming organoids can lead to inconsistent amounts of drug or bacteria in the lumen of each organoid. One elegant solution is the creation of reverse-polarity organoids
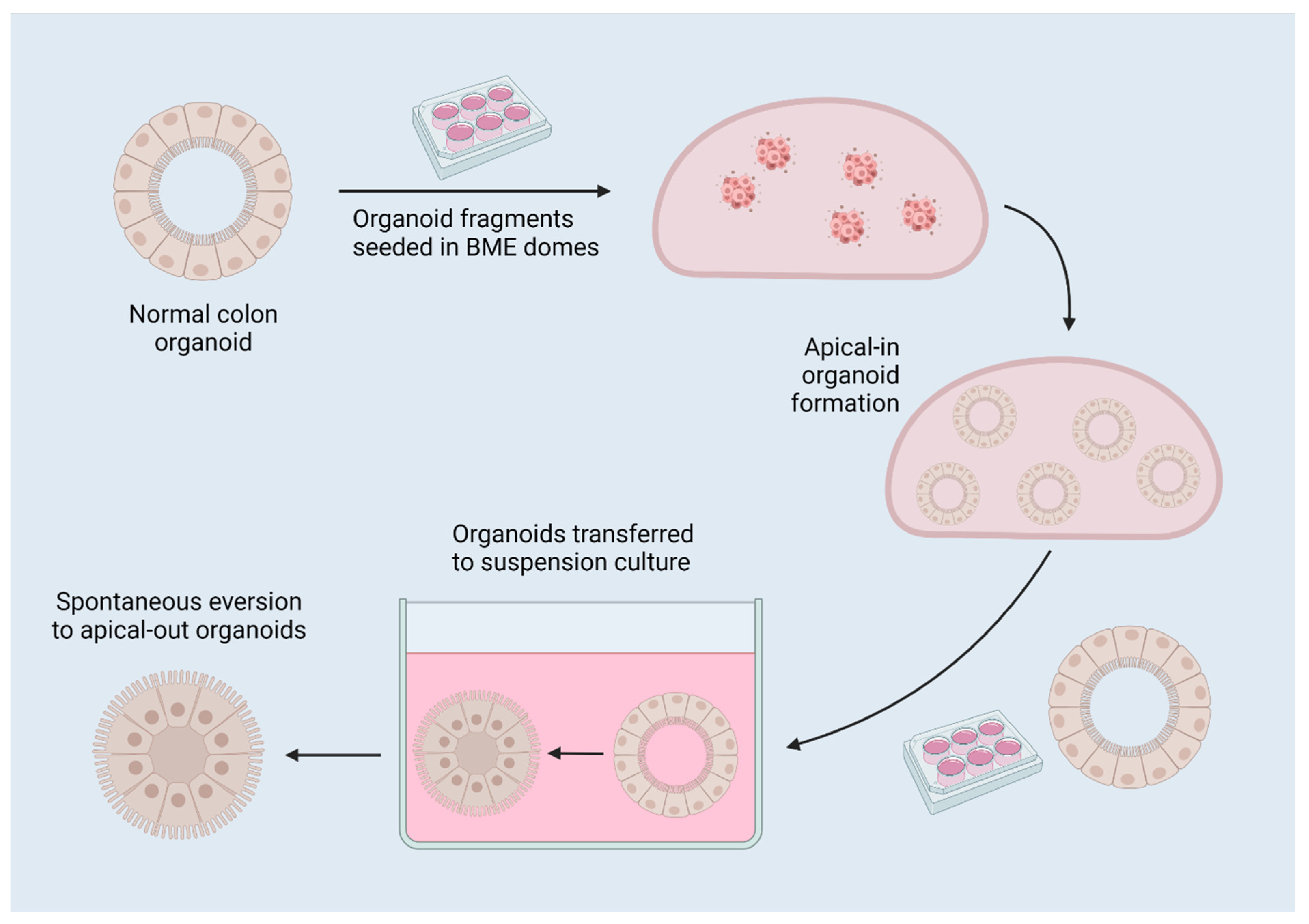
[6][7]. When removed from their extracellular matrix (ECM) dome (Matrigel, Cultrex, etc.) and placed in suspension culture, the typical basal-out organoids spontaneously evert to become apical-out, reverse-polarity organoids (
Figure 3), as described by Co et al.
[6][7]. Apical-out organoids have been proven to retain their barrier integrity and allow increased bacterial invasion compared to basal-out organoids, representing a more relevant model for colonic function, and facilitating the targeting of receptors on the apical/lumenal epithelial surface by pathogens and for drug screening
[6].
Figure 3. A diagram of reverse-polarity colon organoids. Organoids are established in 3D matrix domes, then released for culture in suspension, which induces spontaneous eversion to apical-out organoids due to the removal of interactions with surrounding matrix. Created with
BioRender.com 17 July 2022.
1.4. Cryopreservation and Recovery
Whole organoids or organoid fragments are preferred over an organoid-derived single-cell digest for cryopreservation due to their enhanced viability when thawed
[15]. Similarly, it has been proposed that adding ROCKi to the cryopreservation media increases the viability of thawed organoids
[15]. A common cryopreservation media formula includes 10% DMSO and 20% foetal calf serum in basal medium, or, alternatively, a commercial solution such as CryoStor (StemCell Technologies, Vancouver, BC, Canada) may be used.
2. Applications of Colon Organoids
2.1. Disease Modelling
Organoids are an in vitro version of the tissue from which they are established. As such, they are a useful system for disease modelling. Each tumour-derived organoid is a model of the tumour from which it is produced and can be used for drug testing in the personalised medicine context. They incorporate a range of cell types seen in the original organ, thereby constituting a more robust and physiologically relevant model than more homogenous 2D cell lines, which tend to contain a single dominant cell type that outcompetes the others. The complexity and physiological relevance of such models can be enhanced by co-culture of organoids with other cell types including immune cells, endothelium, fibroblasts, and microbes, often utilising an organ-on-a-chip model, as will be discussed later.
A model system for researching cystic fibrosis in intestinal organoids with and without a mutated CFTR gene has been described, whereby wild-type organoids swell when exposed to forskolin, but those with a CFTR mutation as seen in cystic fibrosis patients do not respond to forskolin
[16]. This model allows drug assays to be performed in order to find compounds that reverse the effects of defective cystic fibrosis transmembrane conductance regulator protein (CFTR) and identify candidates for treating cystic fibrosis
[16].
Colon organoids have also been used to model SARS-CoV-2 infection. COVID-19 infections primarily occur in the respiratory and digestive tracts, and various colon cell types express the ACE2 protein that SARS-CoV-2 utilises to invade cells. Colon organoids are susceptible to SARS-CoV-2 infection, and this can be prevented through treatment with various compounds to identify “entry inhibitors”
[10].
Matano et al.
[17] utilised organoids derived from normal human intestinal tissue to model the initiation of colorectal cancer (CRC). The influence of pathways known to play a role in CRC were elucidated by introducing mutations via CRISPR-Cas9 site-directed mutagenesis. Sequential mutation of APC, SMAD4, TP53, KRAS, and PIK3CA produced organoids with no dependence on Wnt, R-spondin, EGF, Noggin, or A83-01 in the culture media, and that could form tumours when transplanted into immunodeficient mice
[17]. This highlights the potential of organoids as a model for delineating the mechanisms of early tumourigenesis.
The colon air–liquid interface (ALI) model was extensively characterised by Wang et al.
[18], who grew colon spheroids, dissociated them into single cells, and seeded them on Transwell inserts. After being submerged for 7 days in L-WRN conditioned medium, the cells were cultured as an ALI, with the bottom surface of the insert in contact with media and the top surface exposed to air, for 28 days. During this time, the cells differentiated into a heterogenous 2D monolayer consisting of MUC2
+ goblet cells (by day 4), CHGA
+ enteroendocrine cells (day 7), and SLC26A3
+ enterocytes/colonocytes (day 14–21)
[18]. Day 21 ALI cultures could be reformed following passage and recover from cryopreservation, and performed mucosal repair following damage
[18]. These experiments present spheroid- or organoid-derived ALI cultures as a robust in vitro model for colon function, including an intact epithelial barrier, for applications such as co-culture with bacteria
[19] or studies of colonic metabolism
[20].
Colon tumour patient-derived organoids (PDOs) have been co-cultured with stromal cells to simulate the in vivo influence of the tumour microenvironment (TME) in drug response. Dijkstra et al.
[21] developed organoids from mismatch repair-deficient colon tumours, which have higher mutational burden, and isolated peripheral blood from the same patients to establish a co-culture system. Following two weeks of co-culture, levels of tumour-reactive T-cells with specific killing activity increased from undetectable levels in some cases to around 1–3% of the total pool of CD8
+ T cells across samples
[21]. Neal et al.
[22] developed an ALI model utilising tumour tissue samples that had been minced but not digested enzymatically, thereby retaining tumour architecture in terms of associations between tumour cells and stromal cells. The stroma contained fibroblasts, which persisted for up to 4 weeks after ALI culture establishment, and immune cells including tumour-infiltrating lymphocytes and macrophages, which steadily decreased in number over time but could still be detected for 30–60 days and retained the immune cell diversity observed in the parental tissue
[22]. These results lay the groundwork for future organoid investigations that will allow for increased translation of medicines from the lab to the clinic.
2.2. Drug Screening
The use of organoids for drug screening has recently been reviewed by Rae et al.
[23]. For testing an array of compounds on colon tumour organoids in a high-throughput screening (HTS) setting, Boehnke et al.
[4] successfully established organoids in 384-well plates. Tumour organoids were first established in 12-well plates as described above, then digested to a single-cell suspension and seeded in Matrigel domes at a density of 5000 cells per dome. To measure the response of tumour organoids to a range of compounds, the CellTitre-Glo viability assay (Promega) was used to report on ATP consumption as a measure of the metabolic activity of the cells, as a proxy for cell viability
[4]. Toshimitsu et al. reported a similar protocol utilising 384-well plates, but rather than embedding colon tumour cells in solid Matrigel domes, cells were grown in suspension culture in media supplemented with 2% Matrigel
[24]. Organoids formed from this single-cell suspension, and the plates were incubated on a rotating platform. This system greatly enhances the scalability and establishment speed of HTS assays. To ensure that an equal number of cells were seeded per well, they were labelled with GFP and dispensed by a modified FACS system. The GFP-labelled cells allowed organoid growth to be tracked before, during and after drug treatment, and this method of drug effect measurement was comparable to ATP-based luminescence assays
[24]. The results of drug testing performed on the organoids established in a suspension culture based on 2% Matrigel in culture medium were very similar to the results seen in vivo
[24] and may be a useful alternative to the reverse-polarity method described above.
Vlachogiannis et al.
[25] carried out a comprehensive comparison of PDOs with their parental tissues, and utilised these organoids to test the feasibility of genomically-guided personalised treatment. PDOs closely phenocopied the morphology and expression of CDX2 and CK7 seen in the parental tissues
[25]. Furthermore, 96% of mutations detected in the PDOs and tissues were shared
[25]. PDOs were treated with a library of 55 drugs either under investigation in clinical trials or already used clinically. Response to these compounds was directly correlated with genetic alterations to the drug target; for instance, the strongest response to the ERBB2/EGFR inhibitor lapatinib was seen in a PDO with an
ERBB2 amplification, a PDO with an
AKT1 amplification and E17K mutation displayed the greatest response to two AKT inhibitors (MK-2206 and GSK690693), and those PDOs with RB1 amplifications responded to CDK4/CDK6 inhibitor palbociclib
[25]. In vitro PDO models and PDO-xenograft mouse models both replicated the responses to multi-kinase inhibitor regorafenib and ATM/ATR inhibitor VX-970 seen in clinical trials in humans
[25]. Finally, this study shows that organoids reliably replicate both the histopathology of tumours and their response to drugs as seen in patients, suggesting that organoids may be valuable for screening drugs for personalised treatment or to be used in a co-clinical trial context. Similarly, Schnalzger et al.
[26] utilised PDOs and live-cell fluorescence imaging to trial chimeric antigen receptor (CAR) lymphocytes engineered to be tumour-specific, revealing efficient killing of tumour organoids but with accompanying off-target effects on normal colon organoids, highlighting the utility of organoids for testing the safety of new treatments before administration to humans.
Drug response can also be followed by tracking organoid phenotypes over time using microscopy, including organoid size and architecture (solid vs. cystic)
[27]; however, to mitigate the inherent variability between organoids derived from different patients, assays tracking cell death, senescence and growth rate from a baseline value established before treatment would improve the accuracy, success and utility of HTS assays.
This entry is adapted from the peer-reviewed paper 10.3390/organoids2010003