Inflammation is defined as the response of the immune system to a variety of stimuli that might be infectious or tissue harmful. Regardless of the initial insult, there is a series of programmed sequelae depending on the ability of the immune system to eliminate the ‘enemy’ and restore the tissues’ normal structure and function. The inflammatory process can be divided, without clearly defined and therefore overlapping borders, into three sequential phases, including the acute phase, the intermediate and the restore/repair phase. The pivotal role of inflammation in the pathophysiology of heart-failure (HF) development and progression has long been recognized. High blood levels of pro-inflammatory and inflammatory markers are present and associated with adverse outcomes in patients with HF.
- heart failure
- inflammation
- homeostasis
1. Following a Self-Catastrophic Path—Missing the Balance
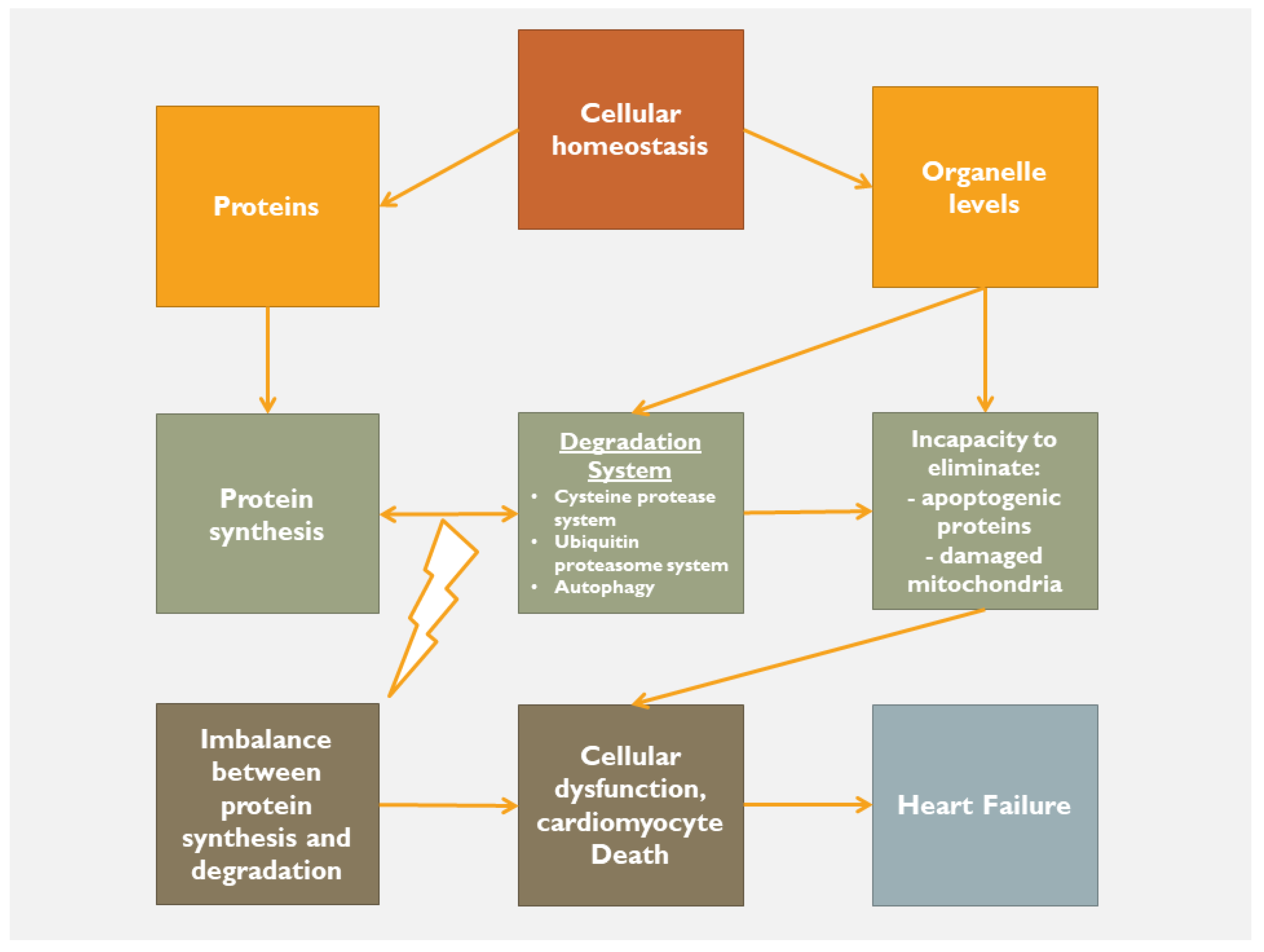
2. Homeostatic Mechanisms
3. Organelle Communication
4. Targeting Inflammation, Oxidative Stress and Mitochondrial Dysfunction
This entry is adapted from the peer-reviewed paper 10.3390/jcdd10010019
References
- DeLuca, G.; Cavalli, G.; Campochiaro, C.; Tresoldi, M.; Dagna, L. Myocarditis: An interleukin-1-mediated disease? Front. Immunol. 2018, 9, 1335.
- Toldo, S.; Kannan, H.; Bussani, R.; Anzini, M.; Sonnino, C.; Sinagra, G.; Merlo, M.; Mezzaroma, E.; De-Giorgio, F.; Silvestri, F.; et al. Formation of the inflammasome in acute myocarditis. Int. J. Cardiol. 2014, 171, e119–e121.
- Butts, B.; Gary, R.A.; Dunbar, S.B.; Butler, J. The importance of NLRP3 inflammasome in heart failure. J. Card. Fail. 2015, 21, 586–593.
- Onódi, Z.; Ruppert, M.; Kucsera, D.; Sayour, A.A.; Tóth, V.E.; Koncsos, G.; Novák, J.; Brenner, G.B.; Makkos, A.; Baranyai, T.; et al. AIM2-driven inflammasome activation in heart failure. Cardiovasc. Res. 2021, 117, 2639–2651.
- Wu, J.; Dong, E.; Zhang, Y.; Xiao, H. The role of the inflammasome in heart failure. Front. Physiol. 2021, 12, 709703.
- Youker, K.A.; Assad-Kottner, C.; Cordero-Reyes, A.M.; Trevino, A.R.; Flores-Arredondo, J.H.; Barrios, R.; Fernandez-Sada, E.; Estep, J.D.; Bhimaraj, A.; Torre-Amione, G. High proportion of patients with end-stage heart failure regardless of aetiology demonstrates anti-cardiac antibody deposition in failing myocardium: Humoral activation, a potential contributor of disease progression. Eur. Heart J. 2014, 35, 1061–1068.
- Adamo, L.; Rocha-Resende, C.; Lin, C.-Y.; Evans, S.; Williams, J.; Dun, H.; Li, W.; Mpoy, C.; Andhey, P.S.; Rogers, B.E.; et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight 2020, 5, e134700.
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435.
- Kologrivova, I.; Shtatolkina, M.; Suslova, T.; Ryabov, V. Cells of the immune system in cardiac remodeling: Main players in resolution of inflammation and repair after myocardial infarction. Front. Immunol. 2021, 12, 664457.
- Isobe, Y.; Kato, T.; Arita, M. Emerging roles of eosinophils and eosinophil-derived lipid mediators in the resolution of inflammation. Front. Immunol. 2012, 3, 270.
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600.
- Stachowski, M.J.; Holewinski, R.J.; Grote, E.; Venkatraman, V.; Van Eyk, J.E.; Kirk, J.A. Phospho-Proteomic Analysis of Cardiac Dyssynchrony and Resynchronization Therapy. Proteomics 2018, 18, e1800079.
- Henao-Mejia, J.; Elinav, E.; Strowig, T.; Flavell, R.A. Inflammasomes: Far beyond inflammation. Nat. Immunol. 2012, 13, 321–324.
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329.
- Chen, Y.-J.; Quintanilla, C.G.; Liou, J. Recent insights into mammalian ER–PM junctions. Curr. Opin. Cell Biol. 2019, 57, 99–105.
- Hwang, J.; Qi, L. Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605.
- Loi, M.; Molinari, M. Mechanistic insights in recov-ER-phagy: Micro-ER-phagy to recover from stress. Autophagy 2020, 16, 385–386.
- Zhou, H.; Wang, S.; Hu, S.; Chen, Y.; Ren, J. ER–mitochondria microdomains in cardiac ischemia–reperfusion injury: A fresh perspective. Front. Physiol. 2018, 9, 755.
- Guido, D.; Demaurex, N.; Nunes, P. Junctate boosts phagocytosis by recruiting endoplasmic reticulum Ca2+ stores near phagosomes. J. Cell Sci. 2015, 128, 4074–4082.
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521.
- Pastor-Cantizano, N.; Ko, D.K.; Angelos, E.; Pu, Y.; Brandizzi, F. Functional diversification of ER stress responses in Arabidopsis. Trends Biochem. Sci. 2020, 45, 123–136.
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947.
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159.
- Reddish, F.N.; Miller, C.L.; Gorkhali, R.; Yang, J.J. Calcium dynamics mediated by the endoplasmic/sarcoplasmic reticulum and related diseases. Int. J. Mol. Sci. 2017, 18, 1024.
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int. J. Mol. Sci. 2017, 18, 1576.
- Annunziata, I.; Sano, R.; d’Azzo, A. Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis. 2018, 9, 328.
- Gao, P.; Yan, Z.; Zhu, Z. Mitochondria-associated endoplasmic reticulum membranes in cardiovascular diseases. Front. Cell Dev. Biol. 2020, 8, 604240.
- Silva-Palacios, A.; Zazueta, C.; Pedraza-Chaverri, J. ER membranes associated with mitochondria: Possible therapeutic targets in heart-associated diseases. Pharmacol. Res. 2020, 156, 104758.
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-associated ER membranes–the origin site of autophagy. Front. Cell Dev. Biol. 2020, 8, 595.
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 2017, 27, 371–385.
- Kornmann, B. The molecular hug between the ER and the mitochondria. Curr. Opin. Cell Biol. 2013, 25, 443–448.
- Lee, S.; Min, K.-T. The interface between ER and mitochondria: Molecular compositions and functions. Mol. Cells 2018, 41, 1000–1007.
- Kho, C.; Lee, A.; Hajjar, R.J. Altered sarcoplasmic reticulum calcium cycling—Targets for heart failure therapy. Nat. Rev. Cardiol. 2012, 9, 717–733.
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259.
- Guerriero, C.J.; Brodsky, J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012, 92, 537–576.
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ sensing: Its role in calcium homeostasis and signaling. Mol. Cell 2017, 66, 780–788.
- Klecker, T.; Böckler, S.; Westermann, B. Making connections: Interorganelle contacts orchestrate mitochondrial behavior. Trends Cell Biol. 2014, 24, 537–545.
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148.
- Rosati, E.; Sabatini, R.; Rampino, G.; DeFalco, F.; DiIanni, M.; Falzetti, F.; Fettucciari, K.; Bartoli, A.; Screpanti, I.; Marconi, P. Novel targets for endoplasmic reticulum stress-induced apoptosis in B-CLL. Blood 2010, 116, 2713–2723.
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194.
- Wang, Y.; Zhang, X.; Wen, Y.; Li, S.; Lu, X.; Xu, R.; Li, C. Endoplasmic reticulum-mitochondria contacts: A potential therapy target for cardiovascular remodeling-associated diseases. Front. Cell Dev. Biol. 2021, 9, 774989.
- Burkewitz, K.; Feng, G.; Dutta, S.; Kelley, C.A.; Steinbaugh, M.; Cram, E.J.; Mair, W.B. Atf-6 regulates lifespan through ER-mitochondrial calcium homeostasis. Cell Rep. 2020, 32, 108125.
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891.
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr. Mol. Med. 2016, 16, 533–544.
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: Mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Heart Fail. 2013, 6, 572–583.
- Ma, M.; Chen, W.; Hua, Y.; Jia, H.; Song, Y.; Wang, Y. Aerobic exercise ameliorates cardiac hypertrophy by regulating mitochondrial quality control and endoplasmic reticulum stress through M2AChR. J. Cell. Physiol. 2021, 236, 6581–6596.
- Prola, A.; Nichtova, Z.; Pires Da Silva, J.; Piquereau, J.; Monceaux, K.; Guilbert, A.; Gressette, M.; Ventura-Clapier, R.; Garnier, A.; Zahradnik, I.; et al. Endoplasmic reticulum stress induces cardiac dysfunction through architectural modifications and alteration of mitochondrial function in cardiomyocytes. Cardiovasc. Res. 2019, 115, 328–342.
- Eisner, V.; Csordás, G.; Hajnóczky, G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle–pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. 2013, 126, 2965–2978.
- Szabo, T.M.; Frigy, A.; Nagy, E.E. Targeting Mediators of Inflammation in Heart Failure: A Short Synthesis of Experimental and Clinical Results. Int. J. Mol. Sci. 2021, 22, 13053.
- Champs, B.; Degboé, Y.; Barnetche, T.; Cantagrel, A.; Ruyssen-Witrand, A.; Constantin, A. Short-term risk of major adverse cardiovascular events or congestive heart failure in patients with psoriatic arthritis or psoriasis initiating a biological therapy: A meta–analysis of randomised controlled trials. RMD Open 2019, 5, e000763.
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T.; Anti-TNF Therapy against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure: Results of the anti-TNF Therapy against Congestive Heart Failure (ATTACH) trial. Circulation 2003, 107, 3133–3140.
- Mann, D.L.; McMurray, J.J.V.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602.
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019, 139, 1289–1299.
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1β inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2020, 41, 2153–2163.
- Kjekshus, J.; Apetrei, E.; Barrios, V.; Böhm, M.; Cleland, J.G.F.; Cornel, J.H.; Dunselman, P.; Fonseca, C.; Goudev, A.; Grande, P.; et al. Rosuvastatin in older patients with systolic heart failure. N. Engl. J. Med. 2007, 357, 2248–2261.
- Tavazzi, L.; Maggioni, A.P.; Marchioli, R.; Barlera, S.; Franzosi, M.G.; Latini, R.; Lucci, D.; Nicolosi, G.L.; Porcu, M.; Tognoni, G.; et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1231–1239.
- Hare, J.M.; Mangal, B.; Brown, J.; Fisher, C.; Freudenberger, R.; Colucci, W.S.; Mann, D.L.; Liu, P.; Givertz, M.M.; Schwarz, R.P.; et al. Impact of oxypurinol in patients with symptomatic heart failure: Results of the OPT-CHF study. J. Am. Coll. Cardiol. 2008, 51, 2301–2309.
- Torre-Amione, G.; Anker, S.D.; Bourge, R.C.; Colucci, W.S.; Greenberg, B.H.; Hildebrandt, P.; Keren, A.; Motro, M.; Moyé, L.A.; Otterstad, J.E.; et al. Advanced Chronic Heart Failure Clinical Assessment of Immune Modulation Therapy Investigators. Results of a non-specific immunomodulation therapy in chronic heart failure (ACCLAIM trial): A placebo-controlled randomised trial. Lancet 2008, 371, 228–236.
- Moreira, D.M.; Vieira, J.L.; Gottschall, C.A.M. The effects of METhotrexate therapy on the physical capacity of patients with ISchemic heart failure: A randomized double-blind, placebo-controlled trial (METIS trial). J. Card. Fail. 2009, 15, 828–834.
- Deftereos, S.; Giannopoulos, G.; Panagopoulou, V.; Bouras, G.; Raisakis, K.; Kossyvakis, C.; Karageorgiou, S.; Papadimitriou, C.; Vastaki, M.; Kaoukis, A.; et al. Anti-inflammatory treatment with colchicine in stable chronic heart failure: A prospective, randomized study. JACC Heart Fail. 2014, 2, 131–137.
- Givertz, M.M.; Anstrom, K.J.; Redfield, M.M.; Deswal, A.; Haddad, H.; Butler, J.; Tang, W.H.W.; Dunlap, M.E.; Le Winter, M.M.; Mann, D.L.; et al. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: The xanthine oxidase inhibition for hyperuricemic heart failure patients (EXACT-HF) study. Circulation 2015, 131, 1763–1771.
- Yokoe, I.; Kobayashi, H.; Kobayashi, Y.; Giles, J.T.; Yoneyama, K.; Kitamura, N.; Takei, M. Impact of tocilizumab on N-terminal pro-brain natriuretic peptide levels in patients with active rheumatoid arthritis without cardiac symptoms. Scand. J. Rheumatol. 2018, 47, 364–370.
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413.