Autophagy is a cellular process that removes damaged components of cells and recycles them as biochemical building blocks. Autophagy can also be induced to protect cells in response to intra- and extracellular stresses, including damage to cellular components, nutrient deprivation, hypoxia, and pathogenic invasion. Dysregulation of autophagy has been attributed to various diseases. In particular, autophagy protects cancer cells by supporting tumor cell survival and the development of drug resistance. The ULK complex is an early-stage regulator of autophagy and attracted particular attention as a drug target. Among ULK isoforms, ULK1, ULK2, ULK3, ULK4, and serine/threonine-protein kinase 36 (STK36), ULK1 have been most extensively studied.
- Unc-51-like autophagy-activating kinase
- autophagy
- cancer
1. Discovery and Development of Unc-51-like Autophagy-Activating Kinase Inhibitors
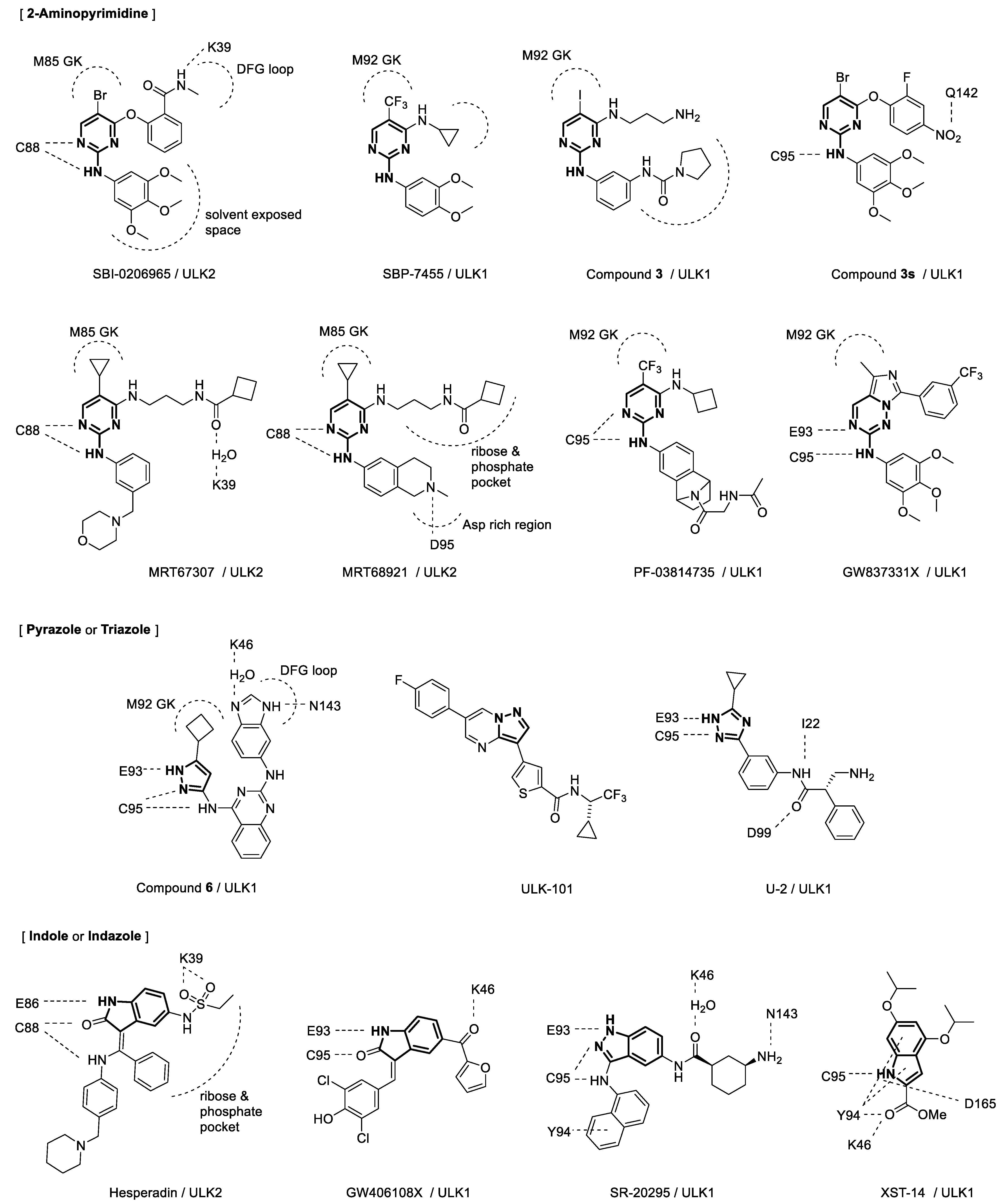
2. Biological and Anticancer Effects of Unc-51-like Autophagy-Activating Kinase Inhibitors
| Compound | IC50 (ULK1) | IC50 (ULK2) | Cancer (Cell Line) | Synergistic Co-Treatment | |
|---|---|---|---|---|---|
| 1 | SBI-0206965 | 108 nM [1] 306 nM [5] 130 nM [2] |
711 nM [1] 3.88 μM [5] |
Non-small cell lung cancer (A549, H460) [1][22] Neuroblastoma (SK-N-AS, SH-SY5Y, SK-N-DZ) [12] Renal cell carcinoma (A498, ACHN) [13] Leukemia (MV4-11, MOLM-13, HL-60, U937) [14][23] Breast cancer (MDA-MB-231, MCF-7) [24] |
mTOR inhibitor AZD8055 [1], daunorubicin [23], doxorubicin [24], cisplatin [22], TRAIL a [12] |
| 2 | SBP-7455 | 13 nM [2] | Triple negative breast cancer (MDA-MB-468, BT549, MDA-MB-231) [2] | PARP inhibitors olaparib, niraparib [2] | |
| 3 | Compound 6 | 8 nM [3] | - | ||
| 4 | Compound 3 | 120 nM [4] | 360 nM [4] | - | |
| 5 | MRT67307 | 45 nM [15] 170 nM [4] 38.2 nM [5] |
38 nM [15] 230 nM [4] 92.3 nM [5] |
Leukemia (THP-1, U939, Molt4, HEL92.1.7, K562, Raji, Jurkat, HL60) [25] | |
| 6 | MRT68921 | 2.9 nM [15] 17.0 nM [5] |
1.1 nM [15] 20.8 nM [5] |
Mesothelioma (M28, REN) [26] b Leukemia (REH, MV4-11, MOLM-13, HL-60, U937, THP-1, Molt4, HEL92.1.7, K562, Raji, Jurkat, HL60) [14][17][25] Ovarian cancer (OVCAR3/4/8, COV318/362, CaOV3) [18] Various tumors [16] |
Carboplatin and pemetrexed [26] |
| 7 | Compound 3s | 99.15% inhibition at 10 μM [8] c | Lung cancer (A549) [8] Lymphoma (U937) [8] Breast cancer (HL60) [8] Acute myeloid leukemia (MDA-MB-469) [8] |
||
| 8 | PF-03814735 | KD 18.1 nM [5] d | KD 58.0 nM [5] d | Solid tumors [19][20] | |
| 9 | Hesperidin | KD 16.8 nM [5] d | KD 47.3 nM [5] d | Various tumors [21] | |
| 10 | SR-20295 | 45 nM [9] | - | ||
| 11 | ULK-101 | 8.3 nM [6] | 30 nM [6] | Osteosarcoma (U2OS) [6] Non-small cell lung cancer (H838, H727, H2030, A549) [6] |
|
| 12 | XST-14 | 13.6 nM [10] | 70.9 nM [10] | Hepatocellular carcinoma (HepG2, Hep3B) [10] | sorafenib [10] |
| 13 | GW837331X | 646 nM [7] | Similar to ULK1 e [7] | - | |
| 14 | GW406108X | 427 nM [7] | Similar to ULK1 e [7] | - | |
| 15 | U-2 | 0.5 μM [11] | Hepatocellular carcinoma (SMMC-7721, HepG2, L02) [11] |
3. Combination Therapy of Unc-51-like Autophagy-Activating Kinase Inhibitors with Other Anticancer Agents
This entry is adapted from the peer-reviewed paper 10.3390/ijms24020953
References
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.-C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297.
- Ren, H.; Bakas, N.A.; Vamos, M.; Chaikuad, A.; Limpert, A.S.; Wimer, C.D.; Brun, S.N.; Lambert, L.J.; Tautz, L.; Celeridad, M.; et al. Design, Synthesis, and Characterization of an Orally Active Dual-Specific ULK1/2 Autophagy Inhibitor that Synergizes with the PARP Inhibitor Olaparib for the Treatment of Triple-Negative Breast Cancer. J. Med. Chem. 2020, 63, 14609–14625.
- Lazarus, M.B.; Novotny, C.J.; Shokat, K.M. Structure of the Human Autophagy Initiating Kinase ULK1 in Complex with Potent Inhibitors. ACS Chem. Biol. 2015, 10, 257–261.
- Lazarus, M.B.; Shokat, K.M. Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorg. Med. Chem. 2015, 23, 5483–5488.
- Chaikuad, A.; Koschade, S.E.; Stolz, A.; Zivkovic, K.; Pohl, C.; Shaid, S.; Ren, H.; Lambert, L.J.; Cosford, N.D.P.; Brandts, C.H.; et al. Conservation of structure, function and inhibitor binding in UNC-51-like kinase 1 and 2 (ULK1/2). Biochem. J. 2019, 476, 875–887.
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience 2018, 8, 74–84.
- Zachari, M.; Rainard, J.M.; Pandarakalam, G.C.; Robinson, L.; Gillespie, J.; Rajamanickam, M.; Hamon, V.; Morrison, A.; Ganley, I.G.; McElroy, S.P. The identification and characterisation of autophagy inhibitors from the published kinase inhibitor sets. Biochem. J. 2020, 477, 801–814.
- Sun, D.; Yang, Z.; Zhen, Y.; Yang, Y.; Chen, Y.; Yuan, Y.; Zhang, L.; Zeng, X.; Chen, L. Discovery of 5-bromo-4-phenoxy-N-phenylpyrimidin-2-amine derivatives as novel ULK1 inhibitors that block autophagy and induce apoptosis in non-small cell lung cancer. Eur. J. Med. Chem. 2020, 208, 112782.
- Wood, S.D.; Grant, W.; Adrados, I.; Choi, J.Y.; Alburger, J.M.; Duckett, D.R.; Roush, W.R. In Silico HTS and Structure Based Optimization of Indazole-Derived ULK1 Inhibitors. ACS Med. Chem. Lett. 2017, 8, 1258–1263.
- Xue, S.T.; Li, K.; Gao, Y.; Zhao, L.Y.; Gao, Y.; Yi, H.; Jiang, J.D.; Li, Z.R. The role of the key autophagy kinase ULK1 in hepa-tocellular carcinoma and its validation as a treatment target. Autophagy 2020, 16, 1823–1837.
- He, S.; Liu, Y.; Li, Q.; Lyu, W.; Feng, F.; Guo, Q.; Zhao, L.; Sun, H. In silico approaches using pharmacophore model combined with molecular docking for discovery of novel ULK1 inhibitors. Future Med. Chem. 2021, 13, 341–361.
- Dower, C.M.; Bhat, N.; Gebru, M.T.; Chen, L.; Wills, C.A.; Miller, B.A.; Wang, H.-G. Targeted Inhibition of ULK1 Promotes Apoptosis and Suppresses Tumor Growth and Metastasis in Neuroblastoma. Mol. Cancer Ther. 2018, 17, 2365–2376.
- Lu, J.; Zhu, L.; Zheng, L.P.; Cui, Q.; Zhu, H.H.; Zhao, H.; Shen, Z.J.; Dong, H.Y.; Chen, S.S.; Wu, W.Z.; et al. Overexpression of ULK1 Represents a Potential Diagnostic Marker for Clear Cell Renal Carcinoma and the Antitumor Effects of SBI-0206965. EBioMedicine 2018, 34, 85–93.
- Hwang, D.Y.; Eom, J.-I.; Jang, J.E.; Jeung, H.-K.; Chung, H.; Kim, J.S.; Cheong, J.-W.; Min, Y.H. ULK1 inhibition as a targeted therapeutic strategy for FLT3-ITD-mutated acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2020, 39, 85.
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383.
- Chen, Y.; Xie, X.; Wang, C.; Hu, Y.; Zhang, H.; Zhang, L.; Tu, S.; He, Y.; Li, Y. Dual targeting of NUAK1 and ULK1 using the multitargeted inhibitor MRT68921 exerts potent antitumor activities. Cell Death Dis. 2020, 11, 712.
- Skah, S.; Richartz, N.; Duthil, E.; Gilljam, K.M.; Bindesbøll, C.; Naderi, E.H.; Eriksen, A.B.; Ruud, E.; Dirdal, M.M.; Simonsen, A.; et al. cAMP-mediated autophagy inhibits DNA damage-induced death of leukemia cells independent of p53. Oncotarget 2018, 9, 30434–30449.
- Schöffski, P.; Jones, S.F.; Dumez, H.; Infante, J.R.; Van Mieghem, E.; Fowst, C.; Gerletti, P.; Xu, H.; Jakubczak, J.L.; English, P.A.; et al. Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase in-hibitor PF-03814735 in advanced solid tumours. Eur. J. Cancer 2011, 47, 2256–2264.
- Singha, B.; Laski, J.; Valdés, Y.R.; Liu, E.; DiMattia, G.E.; Shepherd, T.G. Inhibiting ULK1 kinase decreases autophagy and cell viability in high-grade serous ovarian cancer spheroids. Am. J. Cancer Res. 2020, 10, 1384–1399.
- Sankhe, K.; Prabhu, A.; Khan, T. Design strategies, SAR, and mechanistic insight of Aurora kinase inhibitors in cancer. Chem. Biol. Drug Des. 2021, 98, 73–93.
- Aggarwal, V.; Tuli, H.S.; Thakral, F.; Singhal, P.; Aggarwal, D.; Srivastava, S.; Pandey, A.; Sak, K.; Varol, M.; Khan, A.; et al. Molecular mechanisms of action of hesperidin in cancer: Recent trends and advancements. Exp. Biol. Med. 2020, 245, 486–497.
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458.
- Qiu, L.; Zhou, G.; Cao, S. Targeted inhibition of ULK1 enhances daunorubicin sensitivity in acute myeloid leukemia. Life Sci. 2019, 243, 117234.
- Yu, L.; Shi, Q.; Jin, Y.; Liu, Z.; Li, J.; Sun, W. Blockage of AMPK-ULK1 pathway mediated autophagy promotes cell apoptosis to increase doxorubicin sensitivity in breast cancer (BC) cells: An in vitro study. BMC Cancer 2021, 21, 195.
- Yang, W.; Li, Y.; Liu, S.; Sun, W.; Huang, H.; Zhang, Q.; Yan, J. Inhibition of ULK1 promotes the death of leukemia cell in an autophagy irrelevant manner and exerts the antileukemia effect. Clin. Transl. Med. 2021, 11, e282.
- Follo, C.; Cheng, Y.; Richards, W.G.; Bueno, R.; Broaddus, V.C. Inhibition of autophagy initiation potentiates chemosensitivity in mesothelioma. Mol. Carcinog. 2017, 57, 319–332.
- Zhang, L.; Zhu, Y.; Zhang, J.; Chen, L. Inhibiting Cytoprotective Autophagy in Cancer Therapy: An Update on Pharmacological Small-Molecule Compounds. Front. Pharmacol. 2022, 13, 966012.
- Lee, C.-S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517.