To further ensure their survival, CSCs can also evade the innate immune system in several ways [
18]. Crucially, in glioblastoma, melanoma, and colorectal cancer, they are able to downregulate the expression of major histocompatibility complex (MHC) I and II molecules [
19,
20,
21]. They are also able to convert a subset of immature dendritic cells (DCs) into transforming growth factor (TGF)β-secreting cells, which ultimately leads to the expansion of regulatory T cells (Tregs) in lymphoid organs [
22]. They are also capable of reducing the activity of natural killer (NK) cells in several ways, e.g., by decreasing the expression of natural killer group 2, member D (NKG2D) ligands in glioblastoma and breast cancer [
19,
23]; by decreasing the expression of ligands for NK cell activating receptors such as NKp44, NKp30, NKp46, and CD16 [
24,
25]; or by increasing the expression of NK cell inhibitory receptor ligands [
24,
25].
2. The Mutual Role of TME and CSCs in Immunomodulation and Stem Cell Niche Maintenance
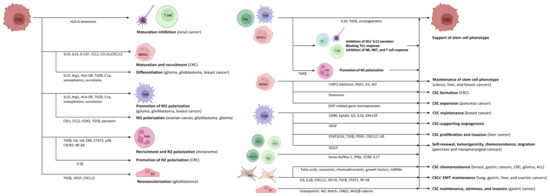
The heterogeneous (i.e., differences in immune cell infiltration and the amount of necrotic tumor cells, interstitial pressure, genetic and epigenetic alterations) and location dependent (i.e., tumor periphery vs. tumor core) tumor microenvironment (TME) is consisting of stroma, extracellular matrix, vasculature, immune cells, and different signaling molecules and pathways (i.e., Notch-, Wnt-, and Hedgehog-pathways) [34,35]. Crosstalk between CSCs and cells in the TME is variable and extensive, involving interconnections between CSCs, tumor stromal cells, and non-CSCs. It is assumed that CSCs inhabit a particular sub-compartment of TME known as the CSC niche. A favorable microenvironment and the absence of specific stimuli that affect cell proliferation keep CSCs quiescent [36]. CSCs survive tumor eradication in quiescence but do not lose their malignant potential, orchestrating the transition to the escape phase. According to acute leukemia studies, repeated tumor growth is triggered by the aggressive and slowly dividing CSC clone [37]. During the escape phase, CSCs secrete cytokines, chemokines, and soluble factors to blunt and alter immune functions and develop immune tolerance in order to create a pro-tumor niche [38]. Tregs, TAMs, and myeloid-derived suppressor cells (MDSCs) are the main organizers of this process, as they mainly enhance the formation of an immune-tolerant TME by secreting interleukin (IL)10, TGFβ, and prostaglandins, as found in colorectal cancer (CRC) [39,40,41,42]. They also inhibit the secretion of IL12 by DCs, block the efficient Th1 response, and inhibit NK, natural killer T (NKT), and effector T cells [40,41,42,43]. During the further development of TME, the formation of angiogenesis-promoting N2-polarized tumor-associated neutrophil granulocytes (TANs) is enhanced through immunosuppressive factors and cytokines (e.g., TGFβ) [44].
In cancer patients, a so-called emergency myelopoiesis is observed, whereby TAMs and MDSCs proliferate in abundance, leading to an abnormal overgrowth of tumor-supporting myeloid cells [45,46]. In addition to the local immune cell dysregulation that occurs in TME, cancers also alter the differentiation of bone marrow progenitors through systemic effects, thereby affecting the extent, composition, and specific functions of hemopoiesis [47,48]. Myeloid cells that have been transferred from the bone marrow to the periphery are transported to the tumor, where they encounter extreme conditions (e.g., hypoxia, low pH, low glucose, and inflammatory signals). The altered microenvironment further enhances their reprogramming towards a pro-tumor phenotype [43,49]. CSCs promote the differentiation of immature myeloid cells by secreting inflammatory mediators (e.g., IL10, IL13) [50]. In addition, granulocyte colony-stimulating factor (G-CSF), C-C motif chemokine ligand (CCL)2, CCL15, and chemokine (C-X-C motif) ligand (CXCL)12 produced by CSCs recruit additional MDSCs to the TME in colon cancers [51,52]. Experimental results in pancreatic cancer have demonstrated that monocyte-derived MDSCs (M-MDSCs) promote CSC expansion and the expression of genes related to the epithelial-to-mesenchymal transition [53]. Similarly, in CRC, granulocyte-derived MDSCs (G-MDSCs) promote CSC formation via exosomes, especially within hypoxic microenvironments [54].
Tregs are also an important component of the TME. Tregs are essentially immunosuppressive and act against tissue damage caused by inflammation [66]. In tumors, however, Tregs suppress the anti-tumor effect of tumor-infiltrating immune cells, thereby promoting tumor escape. The functions of Tregs and their polarization between “anti-tumor” and “pro-tumor” states are regulated by complex molecular and cellular interactions. The binding of semaphorin-4a (Sema-4a) expressed on immune cells to neuropilin-1 (Nrp-1), a receptor for Tregs, enhances the survival and immunosuppressive activity of Tregs. The Nrp-1/Sema-4a pathway is absolutely required for the protection and prolongation of Treg survival in TME [66,67]. Other T cell types can interconvert between phenotypes as well. IL17 producing CD4+ Th17 and Th2 cell are able to switch to IFNγ producing ones via epigenetic, metabolic, and cytokine signaling pathways [68].
Mesenchymal stem cells (MSCs) can promote the chemoresistance of CSCs both directly and indirectly. In breast, colon, and gastric cancers, as well as in glioma, and acute lymphoid leukemia, they contribute to CSC survival during various anti-cancer treatments by secreting fatty acids, exosomes, chemoattractants and growth factors, and by cell–cell contact via miRNA upregulation [
71,
72,
73,
74,
75,
76,
77,
78,
79]. Based on the results in gastric cancer, lung, liver, and ovarian cancers, cancer-associated fibroblasts (CAFs) derived from MSCs, fibroblasts, or epithelial cells also promote EMT and the survival of the CSCs’ stem cell phenotype throughout paracrine actions (IL6, IL1β, and CXCL12 secretion) and via insulin-like growth factor 1 receptor (IGF1R), TGFβ, STAT1, and nuclear factor-κB (NF-kB) pathways [
80,
81,
82,
83,
84,
85].
Besides the positive effects of TME on CSCs, activated CSCs provide favorable conditions for the M2 polarization of TAMs and their pro-tumorigenic effect as well [
40]. Several different factors in gliomas, glioblastomas, and ovarian cancers may play a role in this process, such as periostin (an extracellular matrix protein), colony stimulating factors, CCL2, cyclooxygenase 2 (COX2), or TGFβ [
86,
87,
88,
89,
90]. CSCs contribute to the development of their own vascular network through vascular endothelial growth factor (VEGF) production [
63,
64], and promote their own stem cell development through the activation of Wnt-signaling via the interaction between CSCs and TAMs [
91].
In addition to the action of pro-tumorigenic cytokines via the paracrine pathway, the CSC-derived secretome also plays an important role in the establishment and maintenance of TME. Glioblastoma CSC-derived exosomes can stimulate M2 polarization, programmed death-ligand 1 (PD-L1) expression, and the production of monocyte chemotactic protein 3 (MCP-3) and CXCL1, which promote myeloid cell recruitment, through the STAT3 pathway in glioblastoma [
92]. Using the same secretome in glioma, circulating monocytes produce increased IL10 and arginase 1 (Arg1), decrease Human Leukocyte Antigen DR isotype (HLA-DR) expression, and thereby transform into M-MDSC-like cells [
93]. In breast cancer, exosomes containing TGFβ, complement component 1q (C1q), and semaphorins also promote M2-directed (immunosuppressive) polarization and differentiation of the M-MDSCs [
94]. Maturation of DCs and T cell responses can be inhibited by HLA-G-containing extracellular vesicles, which favor renal tumor cell immune escape mechanisms [
95]. Exosomes of CRC-derived CSCs increase neutrophil granulocyte lifespan and promote the formation of pro-tumorigenic phenotype TANs by increasing the IL1β expression [
96]. Melanoma CSCs can educate neutrophils to support cancer progression in several ways, such as neutrophil recruitment via TGF-β, IL6, and IL8, enhancing N2-polarization by the activation of ERK, STAT3, and P38 pathways, as well as the overexpression of CXCR2 and NF-kB [
97]. MSC-derived secretome of TME also favors the tumorigenic inflammatory response of TAMs by decreasing pro-inflammatory and increasing anti-inflammatory cytokine production [
98].
CSCs may exhibit potent angiogenic properties and contribute to the recruitment of blood vessels during cancer. Vascular endothelial cells (ECs) provide CSCs with supportive signals through cell-to-cell interactions [
105]. Under the impact of TGFβ, glioblastoma CSCs are able to generate pericytes that enable neovascularization and cancer progression [
106]. CSCs produce the angiogenic molecules VEGF and CXCL12 to stimulate EC angiogenesis. ECs, in turn, secrete stemness-maintaining substances such as NO and osteopontin, and stimulate Notch signaling [
107]. Anti-VEGF medication that inhibits ECs can, surprisingly, also be tumorigenic. The anti-VEGF medication may create hypoxia within the TME, which unexpectedly induces VEGF within the TME via a negative feedback loop [
108]. This hypoxic environment can also inhibit CSC differentiation, increase cell treatment resistance, and boost stem-like characteristics in non-CSCs [
108,
109].
Moreover, ECs upregulate capillary morphogenesis gene 2 (CMG2) protein to increase stemness, invasion, and metastasis of CSCs via activating the Wnt/β-catenin pathway observed in gastric cancer [
110] (
Figure 2).
Figure 2. There is an intense association between CSCs and the inflammatory TME. CSCs are able to use immune-competent cells to their own advantage. At the same time, inflammatory TME cells promote the survival of CSCs. Green arrows indicate a stimulatory effect, while the red arrow indicates inhibition. The figure was partly created with
BioRender.com.
3. The CSC-TME Crosstalk in Highly Inflammatory Cancers
The link between inflammation and the development of cancer is rather complex [111]. In the case of acute inflammation following tumor antigen uptake or activation by a Toll-like receptor (TLR) agonist, mature DCs may regulate the anti-tumor immune response by modulating the inflammatory response through various mechanisms (e.g., cross-presentation of tumor antigens and priming of tumor-specific CD8+ T cells, polarization of immune cells towards the anti-tumor phenotype, recruitment of NK cells, thereby maintaining the T cell response) [111,112]. If the acute inflammation is not resolved, it is prolonged over time and transforms into chronic inflammation. In this microenvironment, cancer cells (including CSCs) can hijack DCs, thereby preventing the presentation of tumor antigens, and in addition, they can recruit a variety of immunosuppressive cells (e.g., MDSCs, Tregs, M2-TAMs, and N2-TANs) by producing cytokines, chemokines, and inflammatory mediators. The resulting environment is rich in pro-angiogenic and pro-tumorigenic factors and prevents innate immunity and the T cell response from exerting anti-tumor effects [111,112].
CSC niches have been identified in a number of human cancer types, such as esophageal [114], gastric [115], colorectal [116], liver [117], pancreatic [118], breast [119], ovarian [120], prostate [121], renal [122], brain [123], head and neck [124], lung [125], or melanoma [126]. Numerous studies have examined the similarities and variations between the habitats of various malignancies, as well as the effect of cancer-specific microenvironments on the establishment and growth of CSCs [127,128,129,130,131,132,133].
In the microenvironment of the liver, CSCs promote pro-tumor TME formation in several ways, such as the production of the tissue inhibitor of metalloproteinase 1 (TIMP1) or the activation of the hepatocyte-derived growth factor/hepatocyte-derived growth factor receptor (HGF/HGFR) system by the hypoxia-induced activation of HIF1. Kupffer cells and neutrophils enhance tumor necrosis factor (TNF)α, IL1, and MMP9 production as well [134]. The production of growth factors (e.g., epidermal growth factor receptor /EGFR/, VEGF, PDGF, and stromal cell-derived factor 1 /SDF1/), TNF, and other angiogenic factors also promotes the growth and survival of CSCs and their resistance to radiotherapy and chemotherapy [135,136,137,138]. A number of surface markers are known for CSCs in liver cancer (e.g., CD133, CD90, CD24, CD13, epithelial cell adhesion molecule /EpCAM/, aldehyde dehydrogenase /ALDH/, and hepatic progenitor cell marker OV-6). The expression of stem cell markers confers different properties to CSCs. In CD90+ cells, genes associated with inflammation and drug resistance are upregulated, whereas CD133+ cells are resistant to apoptosis and radiotherapy through activation of the Ak strain transforming/Protein kinase B (Akt/PKB) pathway [139,140].
4. Emergence of CSC Phenotype without TME
Interestingly, the activation of the TLR9 inflammatory signaling pathway, which is part of the innate immune system, can result in the CSC phenotype without the presence of TME. Regarding cell-free DNA (cfDNA), it has been shown that the structure and origin of cfDNA influence its biological effects on cancer cells [
158]. Using an in vitro cellular model that lacked both the TME and the immune system of the tumor-bearing host, scholars investigated the pathobiological effects of self-DNA administration in HT29 colon cancer cells. The scholars provided evidence [
159] for a close existing interplay between TLR9 signaling and the autophagy response, which had significant effects on tumor cell survival in HT29 cells treated with intact or modified self-DNA. Interestingly, it also found colonosphere formation with a strong cytoplasmatic CD133 immunoreactivity in artificially hypermethylated DNA-treated HT29 cells. The scholars further discovered [
160] that the combined use of tumorous self-DNA and IGF1R inhibition displays anti-proliferative properties that can be suppressed by inhibiting TLR9 signaling. Autophagy induced by self-DNA and IGF1R inhibitor also resulted in the survival of CD133-positive HT29 stem-like cancer cells, which may play a role in the CRC recurrence. Since HT29 cancer cells are wildtype regarding Kirsten rat sarcoma virus (K-Ras) mutation, it cannot be ruled out that this observed phenomenon is partly mediated by the RAS/extracellular-signal-regulated kinase (ERK) and phosphoinositide 3-kinase (PI3K)/Akt pathways, with a close connection to the pro-inflammatory factors like IL17, IL22, and IL23 [
161].
5. Utilization of the Inflammatory Process in Cancer Therapy
Theoretically, a number of new ways to boost the immune response against tumors seem to be able to control inflammation caused by cancer.
Local inflammation generated by irradiation or oncolytic viruses can stimulate an anti-cancer innate immune response by activating nucleic acid receptors (TLR9, cyclic GMP–AMP synthase /cGAS/-stimulator of interferon genes /STING/, or RIG-I like receptor /RLR/), followed by a type I IFN response [
158]. The PI3K/Akt/mammalian target of rapamycin (mTOR) pathway is one of the major signaling pathways in CSCs involved in stemness maintenance, proliferation, differentiation, EMT, migration, and autophagy. Therefore, inhibiting the PI3K/Akt/mTOR pathway may also be a promising targeted cancer treatment method [
163].
Restricting the infiltration and function of immunosuppressive cells (e.g., MDSCs, Tregs, M2-TAMs, and N2-TANs) may restore immune surveillance by blocking inflammatory pathways [
111]. Immunotherapeutic strategies such as immune checkpoint blockage, monoclonal antibodies, vaccination, CD8
+ T cell treatment, and activation of innate immune responses such as NK cells, cytokine-induced killer cells, can be used to target CSCs [
164,
165]. Loss of cancer antigen expression, activation of oncolytic pathways, and promotion of an immunosuppressive milieu and (epi)genetic modifications that diminish their identification by the immune system are just a few of the ways that CSCs have developed to evade a potential attack from the immune system. These immunotherapeutic techniques have the potential to increase CSCs’ sensitivity to chemotherapy and/or radiotherapy. Immune checkpoint blockage strategies, for instance, can inhibit the immunosuppressive activity of CSCs and other immunosuppressive cells inside the TME. CSCs and cancer cells generate PD-L1, whereas Tregs express its receptor PD-1 [
107,
166,
167]. CSCs also distribute PD-1 to their respective specialization [
165]. PD-L1 could lead to the depletion and malfunction of effector T cells [
168] and prevent CSCs from evading the immune system. Importance is placed on employing immunotherapeutic strategies targeting specific CSC markers and antigens that are preferentially expressed by the cells [
165].
Besides targeting CAF-derived components, depleting pro-tumor CAFs or transforming them into dormant or anti-tumor cells are all potential anti-cancer therapeutic strategies as well [
174,
175,
176,
177,
178,
179]. The capacity of CAFs to confer stemness to cancer cells renders this treatment possibility intriguing. Compared to epithelial cancer cells, immunological cells, and endothelial cells, CAFs are more positively connected with gene sets associated with poor prognosis, providing support for targeting CAFs as a viable therapeutic route. However, the variety of CAFs needs the identification of more specific markers, as there are CAFs that inhibit tumor growth. Intriguingly, the tumor devoid of myofibroblasts displayed improved spheroid formation, indicating a higher proportion of CSCs. Indeed, the identification of CAF subtypes demonstrates that they promote or inhibit tumor progression in a tissue-dependent way, supporting the necessity for additional research into CAF-specific indicators [
180]. Reeducating pro-tumor CAFs into a state of quiescence or even anti-tumor CAFs is an attractive concept. Given that vitamins (e.g., all-trans retinoic acid or the vitamin D metabolite 1α, 25-dihydroxyvitamin D3) are essential for healthy tissue and their toxicity is relatively lower compared to chemotherapy, reusing vitamin analogs to reconfigure stimulated fibroblasts into a quiescent state may be a clinically viable therapeutic strategy [
177,
178]. Reprogramming the fibroblasts using growth factors, as evidenced by the flexibility of CAFs, is another technique for rewiring the fibroblast population. TGFβ inhibits an IL1-induced phenotype and drives the fibroblast to acquire a myofibroblastic phenotype with less carcinogenesis, including reduced expression of markers supporting cancer stemness, such as IL6 and CXCL12 [
179].
Anti-VEGF resistance causes unsuccessful cancer treatment and recurrence. (Epi)genetic changes cause acquired resistance in cancer cells [
181]. Tumor ECs have epigenetic changes that contribute to anti-angiogenic treatment resistance. Anti-resistance therapy may include many anti-angiogenic substances or anti-angiogenic drugs combined with other treatments. Intussusceptive microvascular formation, vasculogenic mimicry, and vascular co-option are anti-angiogenic treatment resistance mechanisms. Angiogenesis and immune cells interact, which is why anti-angiogenic and immunological checkpoint drugs work so well together. Pan-omics profiling improves clinical outcomes and fights anti-angiogenesis drug resistance [
181].