With rapid economic development, separation and purification processes are used widely in industrial production and daily life, and their requirements are increasing [
1]. However, separating bulk mixtures into pure or purer forms incurs high costs. For example, the current market heavily relies on traditional separation processes and energy-intensive separation methods, such as distillation, which accounts for approximately 10%–15% of global energy consumption [
2]. Note that 10 × 10
7 tons of CO
2 emissions and USD 4 billion in energy costs could be saved annually if more energy-efficient separation approaches were used in the United States alone. Therefore, ever-increasing industrialization requires more efficient, economical, and sustainable separation and purification technologies. Membrane separation techniques are favored because of their advantages, such as low energy consumption, no need for additives, small footprint, and easy scale-up. Additionally, membrane separation techniques are potential candidates to replace traditional thermal-based separation methods. They are powerful tools for solving critical global problems and for developing new industrial processes required for sustainable industrial growth.
Currently, membrane applications mainly involve aqueous media such as wastewater treatment [
3], seawater desalination [
4], and gaseous media such as air separation, hydrogen production, natural gas purification, and CO
2 capture [
5]. The separation/purification of aqueous media using membranes has been studied extensively and applied successfully in various industries, and membranes were applied for gas separation later than for aqueous media. In 1980, Permea (Monsanto) commercialized Prism membranes for industrial applications in separating hydrogen from purge gas streams of ammonia plants, thus marking the beginning of the first large-scale industrial application of gas separation membranes [
6]. Since then, membranes for gas separation have attracted considerable attention from industry and academia, and membrane-based gas separation has exponentially grown.
Compared to polymeric membranes, inorganic membranes have many advantages, such as high chemical, thermal, and mechanical stability. Inorganic membranes have been fabricated from crystalline materials such as zeolites, metal-organic frameworks (MOFs), and amorphous materials. In particular, amorphous inorganic membranes, such as silica, carbon molecular sieves, and silicon carbide-based membranes, also feature better control of pore size and size distribution, as summarized in Table 1. This pore-size control allows for better control of permeability and selectivity.
2. Fabrication of SiC Membranes
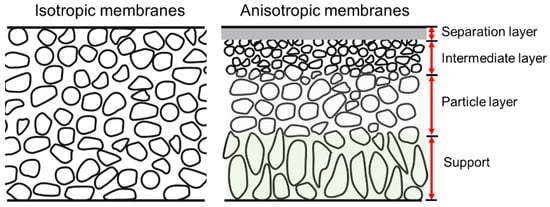
SiC membranes can be divided according to their morphological construction into symmetrical and asymmetrical types, as shown in
Figure 2 [
38]. Relatively high permeation fluxes can be obtained via asymmetric membranes, which consist of a support, transition layers (e.g., a particle layer and/or an intermediate layer), and a separation layer. The supports without separation layers, also known as membrane substrates, play an important role in producing defect-free and reproducible separation layers, which in turn play an important role in meeting industrial demand. The membrane substrates should have good characteristics to provide high flux, porosity, and mechanical strength. Currently, materials used as substrates are mainly alumina, mullite, titania, zirconia, SiC, and their composites [
35,
39]. Among them, SiC is a promising material for fabricating membrane substrates due to its unique properties. Generally, the same SiC substrates can be used as membrane filters, e.g., for microfiltration (MF, 0.05–10 μm), ultrafiltration (UF, 2–50 nm), and nanofiltration (NF, ≤2 nm) [
37,
40,
41]. SiC supports/filters with large pores (>10 μm) are commonly used for air filtering, soot filtering, and catalyst distributors in diesel engines [
37,
42]. Processing methods for these supports/filters, such as extrusion [
43], tape casting [
44,
45], slip-casting [
46], and compression molding [
47], have been extensively investigated.
Figure 2. Schematic of isotropic and anisotropic membranes [
38].
SiC membrane filters usually consist of multiple layers on macroporous supports. Depending on their application purpose, the layers can be designed to generate an upper layer with suitable pore sizes. SiC membrane filters are used mainly in pressure-driven processes, such as MF, UF, and NF, and they have found applications in water treatment, food, and gas cleaning industries. There are a variety of fabrication processes for the fabrication of SiC membrane filters with respect to support characteristics, the range of combinations of raw materials (such as SiC powder compositions, particle sizes, polymer precursors, and sintering additives), deposition techniques, thermal treatment, and applications, as listed in Table 2. The thickness of the top layers ranged from 7 to 125 µm, and the layers were sintered at temperatures up to 2250 °C (Table 2). It is worth noting that the thickness of the top layers (7–19 µm) with transition layers (intermediate layers) was considerably thinner than the top layers directly produced on supports because the transition layers prevent the top-layer solutions from permeating the supports. Additionally, the use of preceramic precursors (e.g., allyl-hydridopolycarbosilane, AHPCS) reduced the thermal treatment temperature significantly to 750 °C, reducing the SiC membrane production cost.
Table 2. Fabrication process and properties of representative SiC membrane filtrations (MF and UF).
| Shape and Pore Size of Supports |
Raw Materials of Layers on Supports |
Layer Deposition Method |
Top Layer Thickness [μm] |
Thermal Treatment |
Pore Size (Type: MF, UF) |
Applications and Other Remarks |
Ref. |
| Transition Layer |
Top Layer |
| Disk, 15 μm |
na. |
α-SiC powder (10 μm), SiC whisker, methylcellulose (MC) 2, CaO 5, ZrO2 5, mullite 5, TL-56NQ 4, water 1 |
Spray coating |
125 |
1150–1250 °C, 2 h in air; then 1350–1500 °C, 4 h in Ar |
2.31 μm (MF) |
-
Hot gas filtration (high permeability, dust removal efficiency, 99.95%)
-
Excellent corrosion resistance in both H2SO4 and NaOH
-
Excellent thermal shock resistance
|
[48] |
| Flat tube, 1.8 μm |
na. |
SiC powder (0.55 μm), IPA 1, PVA 3, PEG 3, Darvan-CN 2, water 1 |
Dip-coating |
12–30 |
900–1300 °C, 1 h |
75–155 nm (MF) |
|
[49] |
| Flat tube, 34.92 μm |
na. |
SiC powder (22 µm), B4C 5, PVA 3, TMAOH 2, water |
Dip-coating |
~100 |
2200–2250 °C |
9.93 μm (MF) |
|
[50] |
| Tube, 15 μm |
na. |
α-SiC powder (0.4 μm and 0.6 μm), Al(NO3)3·9H2O 5, Optapix CS-76 3, polysaccharide dicarbonic acid polymer 3, water 1 |
Dip-coating |
27.3–29.4 |
1600–1900 °C |
0.35 μm (MF) |
-
Oily wastewater treatment (remarkable separation performance)
-
Multi-channeled tubular membranes
-
High chemical and mechanical stability
|
[51] |
| Flat sheet, 5.6–14.1 μm |
na. |
SiC powders (0.5 µm and 3 µm), PAA 2, CMC 3, water 1 |
Dip-coating |
60 |
1900–2000 °C in vacuum |
0.5 μm (1900 °C);
1.4 μm (2000 °C) (MF) |
-
Gas filtration
-
Homogeneous structure
|
[52] |
| Flat disk, <100 μm |
Consisting of several SiC layers;
pore diameter <300 nm |
α-SiC powder (0.4 µm), AHPCS 6, hexane 1, hexane/tetradecane 1 |
Dip-coating |
10–19 |
200 °C, 1 h; 400 °C, 1 h; and then 750 °C, 2 h |
<50 nm (UF) |
|
[53] |
| Tube/na. |
SiC powder (0.6 µm), acetone; pore diameter =130 nm |
PS 7, toluene 1, AHPCS 6, hexane 1 |
Slip-casting
+
dip-coating |
7 |
200 °C, 1 h, 400 °C, 1 h, and then 750 °C, 2 h in Ar; 450 °C, 2 h in air |
Nanoporous SiC membranes |
|
[54] |
Currently, commercialized SiC membranes are mainly used in MF and UF applications. Regardless of the manufacturer, commercial SiC membranes with high permeability (or flux) and good rejection are used widely in water treatment, especially for MF applications [
35,
55]. Although the commercial-scale production of SiC membranes is already a mature technology, manufacturing membranes with smaller pore sizes and narrower pore size distributions is a major challenge, such as for water treatment via NF and RO, and particularly for gas separation using microporous SiC membranes. In recent decades, extensive research has been conducted on the preparation of microporous SiC membranes with productive achievements. The subsequent sections focus on the two main precursor/polymer-derived ceramic approaches for fabricating SiC membranes, namely CVD/CVI and the pyrolysis of preceramic precursors.
2.1. Chemical Vapor Deposition/Infiltration Route
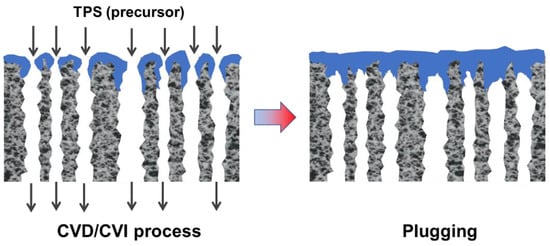
CVD/CVI involves exposing a heated substrate to one or more volatile precursors (e.g., organosilica) that react or decompose on the substrate surface (or on the pore walls) to deposit the desired thin film [
56,
57]. CVD/CVI is a versatile process for manufacturing coatings/films, fibers, powders, and monolithic components [
58]. During the formation of SiC membranes, a schematic diagram of the phenomena that could occur via the CVD/CVI deposition process is shown in
Figure 3. The precursor (e.g., triisopropylsilane, TPS) is carried by the carrier gas (e.g., Ar) into the pore space of the support, where it reacts to form SiC on the pore surface. SiC gradually shrinks/plugs the pores (especially the macropores) and forms a dense membrane layer [
59].
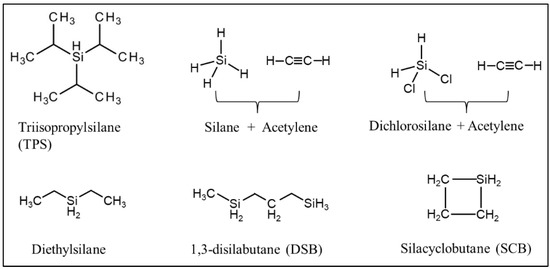
Table 3 lists the representative processing parameters of the SiC membranes obtained by CVD/CVI techniques using the typical precursors shown in
Figure 4. It is worth noting that the vast majority of support materials used to fabricate inorganic membranes, as well as SiC membranes, are α-Al
2O
3. This is due mainly to the ease of commercialization and high economic competitiveness of α-Al
2O
3.
Figure 3. Schematic of SiC membrane preparation via the CVD/CVI deposition process. Adapted from [
59] with permission from Elsevier.
Figure 4. Structures of typical precursors used in CVD/CVI techniques.
Table 3. Representative processing parameters of SiC membranes obtained by CVD/CVI techniques.
| Membranes |
Precursor |
Supports |
Deposition Temperature |
Ref. |
| SiCN |
SiH4/C2H2/NH3 |
α-Al2O3; disk |
1050 °C in Ar |
[60] |
| SiCO |
SiH2Cl2/C2H2/H2 |
γ-Al2O3/α-Al2O3; tube |
800–900 °C in H2 |
[61,62] |
| SiC |
Triisopropylsilane (TPS) |
SiC; disk; and tube |
760–800 °C in Ar/He |
[59] |
| SiC |
Silacyclobutane (SCB) |
Ni-γ-Al2O3/α-Al2O3; tube |
515 °C in Ar |
[63] |
| SiC |
1,3-disilabutane (DSB); TPS |
α-Al2O3; γ-Al2O3/α-Al2O3; tube |
TPS: 700–800 °C in He, and annealed at 1000 °C;
DSB: 650–750 °C in He |
[64,65] |
In an early effort, Hong et al. used the SiH
4/C
2H
2/NH
3 reaction system and α-Al
2O
3 supports to fabricate SiC membranes using hot-wall CVD [
60]. SiC-Si
3N
4 nanoparticles were formed in the gas phase at 1050 °C and deposited in/on the support macropores to form a separation layer with an average pore size of 0.21 μm, which indicates that membrane pore sizes were still in the MF range. Takeda et al. reported the preparation of SiC membranes on γ-Al
2O
3-coated tubular α-Al
2O
3 supports via CVI using the SiH
2Cl
2/C
2H
2/H
2 reaction system at 800–900 °C [
62]. The final SiC membranes exhibited H
2 permeances of 1 × 10
−8 mol/(m
2 s Pa) with an H
2/N
2 selectivity of 3.36 (Knudsen selectivity of 3.74) at 350 °C. Sea et al. reported pure SiC membranes prepared using triisopropylsilane (TPS) on the macropores of tubular α-Al
2O
3 supports using CVD at 700–800 °C, followed by calcination in Ar at 1000 °C [
64]. However, these membranes exhibited a Knudsen-type diffusion mechanism without selectivity toward H
2.
Ciora and Tsotsis et al. were the first to report the fabrication of truly microporous SiC membranes on γ-Al
2O
3-coated tubular α-Al
2O
3 supports via a CVD/CVI technique using two different precursors (i.e., 1,3-disilabutane (DSB) and TPS) [
65]. In their study, the TPS-derived membranes with He permeance ranging from 8.06 × 10
−8 to 1.72 × 10
−6 mol/(m
2 s Pa) and He/N
2 selectivity ranging from 4 to >100 at 550 °C were hydrothermally stable in the presence of high-pressure (1–3 bar) steam. However, DSB-derived versions with He permeance of 3.5 × 10
−7 mol/(m
2 s Pa) and He/N
2 selectivity of ~55 at 550 °C were not. The preparation procedure of membranes using TPS involved multiple steps that required post-treatment for further structural tailoring at temperatures reaching 1000 °C, which increases the cost. Nagano et al. successfully synthesized a helium-selective SiC membrane on the outer surface of a γ-Al
2O
3-coated tubular α-Al
2O
3 support by the pyrolysis of polycarbosilane (PCS) at 800 °C under Ar and then modified by CVI using the SiH
2Cl
2/C
2H
2/H
2 reaction system [
61]. Compared to the original membrane, the membrane modified by CVI exhibited increased He/CO
2 selectivity from 7.7 to 64 and increased He/H
2 selectivity from 1.1 to 4.4. However, the He permeance significantly decreased from 8.9 × 10
−7 to 7.7 × 10
−8 mol/(m
2 s Pa) at 600 °C. The authors attributed this to the densification of the Si-C network and the plugging of surface defects during CVI modification.
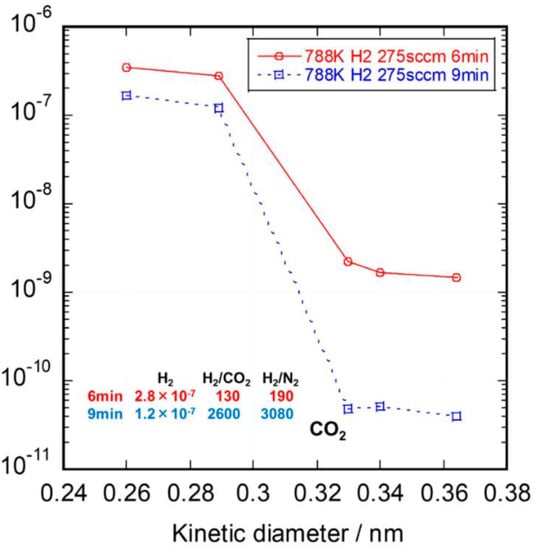
Most recently, Nagano et al. synthesized amorphous SiC membranes on Ni-γ- Al
2O
3/Al
2O
3 supports by counter-diffusion CVD at 515 °C using silacyclobutane and Ar as the carrier gas [
63]. As shown in
Figure 5, the membrane possessed an H
2 permeance of 1.2 × 10
−7 mol/(m
2 s Pa) and excellent H
2/CO
2 selectivity of 2600 at 400 °C at a deposition time of 9 min. The SiC layers were formed within minutes by the CVD/CVI deposition process, which improves the production efficiency of SiC membranes significantly from an industrial point of view. However, as mentioned above, a major issue that remains to be resolved is that SiC membranes formed using CVD/CVI deposition techniques possess dense structures, resulting in low permeability as well as high selectivity suitable for small-molecule gas separation, such as He/H
2 and H
2/CO
2. However, such membranes may not be suitable for separating small-to-mid-sized molecules from larger ones.
Figure 5. Single-gas permeance of SiC membranes formed by the CVD deposition process. Reprinted from [
63] with permission.
2.2. Pyrolysis of the Polymeric Precursor Route
Another well-known method for preparing SiC membranes is the pyrolysis of coated polymeric precursors. The greatest advantage of this method over the CVD/CVI technique is its processing simplicity, low thermal treatment (pyrolysis) temperatures (≤850 °C), and the fact that production can be carried out either continuously or in a batch process. Furthermore, the preceramic polymeric precursor approach features significant advantages in manufacturing membranes with controlled properties and structures, such as composition, hydrophilic/hydrophobic properties, and pore size, which ultimately affect SiC membrane performance.
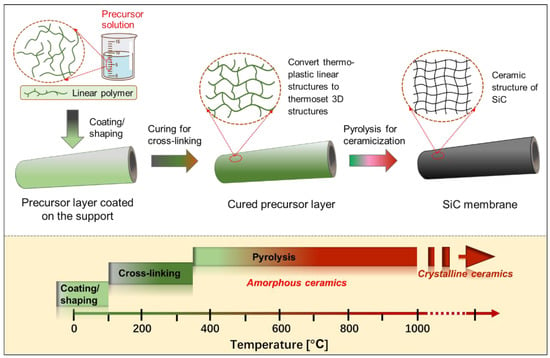
Figure 6 presents a general schematic of the pyrolysis of the preceramic precursor for manufacturing SiC membranes. In particular, the employed polymeric precursors generally undergo three steps using this route to form SiC membranes, depending on the temperature (these steps are shown at the bottom of
Figure 6): coating/shaping, curing for cross-linking, and pyrolysis under an inert atmosphere to complete the polymer-to-ceramic conversion [
66]. In this multi-step process, each step is important for obtaining high-quality membranes.
Figure 6. Schematic of pyrolysis for the preceramic precursor route to manufacture SiC membranes. These steps are also temperature dependent and are shown at the bottom.
2.2.1. Si-Containing Precursors
The fabrication of SiC ceramics by the pyrolysis of a polymeric precursor process is strongly influenced by the chemistry and architecture of the Si-based preceramic precursors, their processing routes, and the parameters used for their pyrolysis. To be suitable for producing SiC ceramic materials, Si-based polymers must meet the following important requirements: (i) the polymers should have a sufficiently high molecular weight to avoid volatilization of components with low molecular weights during subsequent curing and pyrolysis processes; (ii) they should have appropriate rheological characteristics, as well as solubility for the coating/shaping process; (iii) they should have latent reactivity, provided by the presence of specific functional groups (e.g., Si–H, N–H, Si–OH, and Si–CH=CH2), which induce cross-linking during the curing process upon exposure to thermal stimuli, chemical stimuli, or irradiation (e.g., UV, electro-beam, and γ-ray).
2.2.2. Coating/Shaping
The quality of the coating/shaping depends on the deposition techniques and the solution properties of the precursors. Precursor coating methods mainly include dip-coating [31], wipe-coating [55], casting [54], spin-coating [80], spray-coating [48], and even three-dimensional (3D) printing [81]. Dip-coating and wipe-coating are the most frequently used methods for fabricating membranes because of their relative simplicity. An important factor affecting the coating layers is the viscosity (a rheological characteristic) of the coating solutions, which should be controlled carefully by changing the molecular architectures of the precursors, concentration, and solvent species to achieve coating layers with high quality. This is because the viscosity of the coating solutions could affect the thickness and uniformity of the membrane layers.
2.2.3. Curing and Pyrolysis Processes
Curing for Cross-Linking
The coated precursor typically requires to be cured at low temperatures (up to 300–400 °C) prior to pyrolysis [71,83], which plays an important role in determining the final quality of SiC ceramic materials, including their microstructural properties. The curing process of polymeric precursors for cross-linking converts the thermoplastic polymers into thermosetting polymers via a series of reactions, such as dehydrogenation and oxidation, which prevents the coating layers/shapes from fusing together during pyrolysis [84]. Additionally, curing is known to promote high ceramic yields of polymeric precursors because cross-linking prevents the volatilization of precursor components with low molecular weight at high temperatures. To date, several curing techniques have been carried out to produce SiC membranes, such as ultraviolet (UV) radiation, electron beam (EB)/γ-ray irradiation [85,86], and conventional thermal treatment under an oxidizing or inert atmosphere [73,87]. These curing techniques induce various condensation (dehydrogenation or demethanization) and addition reactions converting linear polymer networks to 3D polymer networks.
Pyrolysis for Polymer-To-Ceramic Conversion
After curing for cross-linking, further thermal treatment at elevated temperatures (usually ≥300 °C) and under an inert atmosphere, i.e., pyrolysis, results in an organic-to-inorganic conversion (from thermoset polymers to amorphous SiC ceramics) [66,73]. This conversion is caused mainly by radicals, condensation (dehydrogenation and demethanization), and rearrangement reactions, which lead to the cleavage of chemical bonds and the formation of new bonds accompanied by the elimination of organic groups and the release of gases, such as H2, CH4, and C6H6 [55]. For most polymeric precursors, the conversion from polymeric precursor to amorphous ceramic is complete at <900 °C, followed by crystallization from the amorphous phase at higher temperatures (>1100 °C) and resulting in phase separation [34,55,71,72,73,82].
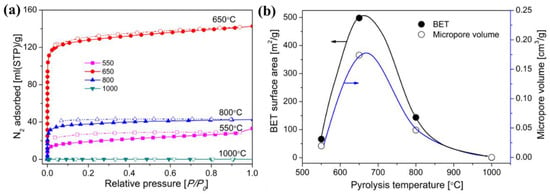
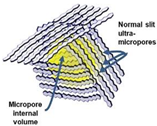
During the organic-to-inorganic ceramic transformation, the microstructure of polymeric precursors undergoes dramatic changes with increasing pyrolysis temperature [55]. As shown in Figure 7, the microporous properties and pore structure parameters (BET and micropore volume) of the pyrolytic precursor (polytitanocarbosilane, TiPCS) powders were analyzed by N2 adsorption–desorption isotherms [55]. The adsorption capacity, micropore volume, and BET surface area increased by firing from 500 °C to 650 °C and then decreased with increasing pyrolysis temperature from 650 °C to 1000 °C. It is worth noting that this is a general conclusion because PCS [72,96], polydimethylsilane (PMS) [97], and AHPCS [73] precursors follow the same trends within a similar pyrolysis temperature range of 300–850 °C. These trends suggest that the decomposition of organic groups generates many micropores at moderate temperatures and the micropores are then narrowed gradually as the pyrolysis temperature increases because of densification and rearrangement reactions.
Figure 8. (a) N2 adsorption–desorption isotherms at 77 K, (b) BET surface area and micropore volume (at a relative pressure of P/P0 = 0.01, pore size ≤ 1 nm) of pyrolyzed precursor (TiPCS) powders. Adapted from [55] with permission from Elsevier.
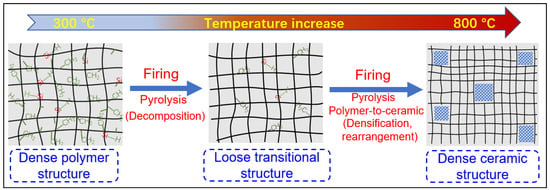
The evolution of the network structure in polymeric precursors pyrolyzed at various temperatures is schematically illustrated in
Figure 8. The evolution of the network structure starts with a dense polymer structure that passes through a loose transitional structure and is then transformed into a relatively denser ceramic structure [
55,
71,
72,
73]. It should be noted that this trend would ultimately affect the gas separation performance of membranes.
Figure 8. Schematic of the evolution of the network structure for polymeric precursors (e.g., AHPCS) pyrolyzed at different temperatures. Reprinted from [
73] with permission from Elsevier.
 [12]
[12]