Reactive oxygen species (ROS) have been described to induce a broad range of redox-dependent signaling reactions in physiological conditions. Nevertheless, an excessive accumulation of ROS leads to oxidative stress, which was traditionally considered as detrimental for cells and organisms, due to the oxidative damage they cause to biomolecules. During ageing, elevated ROS levels result in the accumulation of damaged proteins, which may exhibit altered enzymatic function or physical properties (e.g., aggregation propensity). Emerging evidence also highlights the relationship between oxidative stress and age-related pathologies, such as protein misfolding-based neurodegenerative diseases (e.g., Parkinson’s (PD), Alzheimer’s (AD) and Huntington’s (HD) diseases).
- neurodegenerative diseases
- reactive oxygen species (ROS)
- AD
1. Introduction
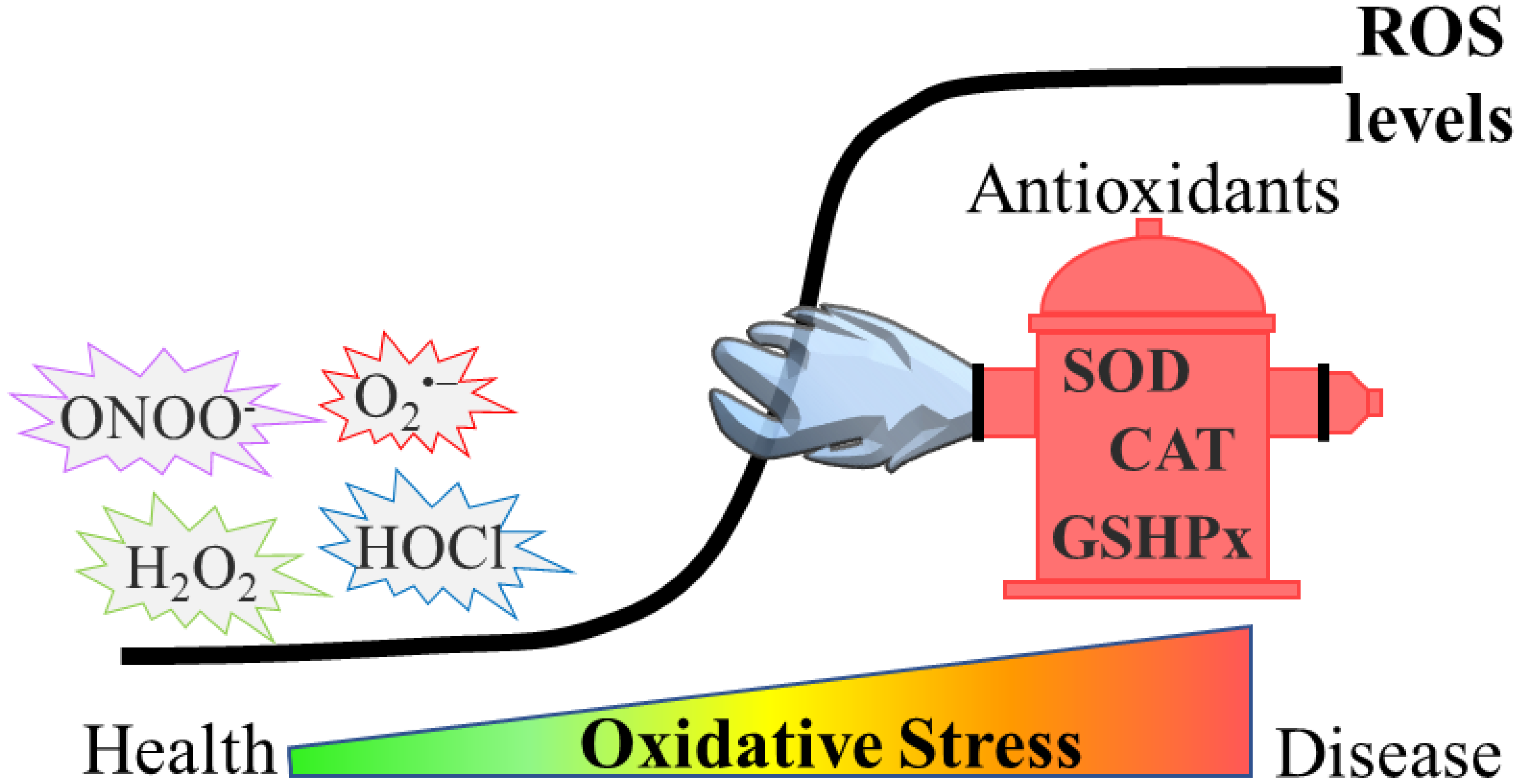
2. Oxidative Stress in Ageing and Age-Related Neurodegenerative Diseases
This entry is adapted from the peer-reviewed paper 10.3390/antiox12010131
References
- Lambeth, J.D. NOX Enzymes and the Biology of Reactive Oxygen. Nat. Rev. Immunol. 2004, 4, 181–189.
- Holmström, K.M.; Finkel, T. Cellular Mechanisms and Physiological Consequences of Redox-Dependent Signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421.
- Ray, S.; Abugable, A.A.; Parker, J.; Liversidge, K.; Palminha, N.M.; Liao, C.; Acosta-Martin, A.E.; Souza, C.D.S.; Jurga, M.; Sudbery, I.; et al. A Mechanism for Oxidative Damage Repair at Gene Regulatory Elements. Nature 2022, 609, 1038–1047.
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for Measuring Reactive Oxygen Species and Oxidative Damage in Cells and in Vivo. Nat. Metab. 2022, 4, 651–662.
- Dickinson, B.C.; Chang, C.J. Chemistry and Biology of Reactive Oxygen Species in Signaling or Stress Responses. Nat. Chem. Biol. 2011, 7, 504–511.
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709.
- Scudamore, O.; Ciossek, T. Increased Oxidative Stress Exacerbates α-Synuclein Aggregation In Vivo. J. Neuropathol. Exp. Neurol. 2018, 77, 443–453.
- Wolschner, C.; Giese, A.; Kretzschmar, H.A.; Huber, R.; Moroder, L.; Budisa, N. Design of Anti- and pro-Aggregation Variants to Assess the Effects of Methionine Oxidation in Human Prion Protein. Proc. Natl. Acad. Sci. USA 2009, 106, 7756–7761.
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 Aggregation Induced by Oxidative Stress Causes Global Mitochondrial Imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142.
- van Dam, L.; Dansen, T.B. Cross-Talk between Redox Signalling and Protein Aggregation. Biochem. Soc. Trans. 2020, 48, 379–397.
- Chen, X.; Guo, C.; Kong, J. Oxidative Stress in Neurodegenerative Diseases. Neural Regen. Res. 2012, 7, 376.
- Park, S.; Park, S.K. Anti-Oxidant and Anti-Aging Effects of Phlorizin Are Mediated by DAF-16-Induced Stress Response and Autophagy in Caenorhabditis Elegans. Antioxidants 2022, 11, 1996.
- Cui, W.B.; Zhang, Z.P.; Bai, X.; Wang, S.S.; Chen, X.H.; Liu, X.; Su, P.J.; Zhi, D.J.; Fei, D.Q.; Zhang, Z.X.; et al. Cryptotanshinone Alleviates Oxidative Stress and Reduces the Level of Abnormally Aggregated Protein in Caenorhabditis Elegans AD Models. Int. J. Mol. Sci. 2022, 23, 10030.
- Wink, M.; Duangjan, C.; Zhang, S.; Song, X.; Wang, X. Exendin-4 Alleviates β-Amyloid Peptide Toxicity via DAF-16 in a Caenorhabditis Elegans Model of Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 955113.
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative Stress and Parkinson’s Disease. Front. Neuroanat. 2015, 9, 91.
- Morales-Martínez, A.; Martínez-Gómez, P.A.; Martinez-Fong, D.; Villegas-Rojas, M.M.; Pérez-Severiano, F.; Del Toro-Colín, M.A.; Delgado-Minjares, K.M.; Blanco-Alvarez, V.M.; Leon-Chavez, B.A.; Aparicio-Trejo, O.E.; et al. Oxidative Stress and Mitochondrial Complex I Dysfunction Correlate with Neurodegeneration in an α-Synucleinopathy Animal Model. Int. J. Mol. Sci. 2022, 23, 11394.
- Aoyama, T.; Peters, J.M.; Iritani, N.; Nakajima, T.; Furihata, K.; Hashimoto, T.; Gonzalez, F.J. Altered Constitutive Expression of Fatty Acid-Metabolizing Enzymes in Mice Lacking the Peroxisome Proliferator-Activated Receptor α (PPARα). J. Biol. Chem. 1998, 273, 5678–5684.
- Cuttler, K.; de Swardt, D.; Engelbrecht, L.; Kriel, J.; Cloete, R.; Bardien, S. Neurexin 2 p.G849D Variant, Implicated in Parkinson’s Disease, Increases Reactive Oxygen Species, and Reduces Cell Viability and Mitochondrial Membrane Potential in SH-SY5Y Cells. J. Neural Transm. 2022, 129, 1435–1446.
- Sasazawa, Y.; Souma, S.; Furuya, N.; Miura, Y.; Kazuno, S.; Kakuta, S.; Suzuki, A.; Hashimoto, R.; Hirawake-Mogi, H.; Date, Y.; et al. Oxidative Stress-Induced Phosphorylation of JIP4 Regulates Lysosomal Positioning in Coordination with TRPML1 and ALG2. EMBO J. 2022, 41, e111476.
- Abubakar, M.B.; Sanusi, K.O.; Ugusman, A.; Mohamed, W.; Kamal, H.; Ibrahim, N.H.; Khoo, C.S.; Kumar, J. Alzheimer’s Disease: An Update and Insights Into Pathophysiology. Front. Aging Neurosci. 2022, 14, 234.
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 57.
- Peng, Y.; Zhang, L.; Zhou, F.; Wang, Y.; Zhang, X.; Fan, J.; Li, S.; Li, X.; Li, Y. Scavenging Reactive Oxygen Species Decreases Amyloid-β Levels via Activation of PI3K/Akt/GLUT1 Pathway in N2a/APP695swe Cells. J. Alzheimers Dis. 2022, 90, 185–198.
- Villegas, L.; Nørremølle, A.; Freude, K.; Vilhardt, F. Nicotinamide Adenine Dinucleotide Phosphate Oxidases Are Everywhere in Brain Disease, but Not in Huntington’s Disease? Front. Aging Neurosci. 2021, 13, 736734.
- Smatlikova, P.; Askeland, G.; Vaskovicova, M.; Klima, J.; Motlik, J.; Eide, L.; Ellederová, Z. Age-Related Oxidative Changes in Primary Porcine Fibroblasts Expressing Mutated Huntingtin. Neurodegener. Dis. 2019, 19, 22–34.
- Machiela, E.; Jeloka, R.; Caron, N.S.; Mehta, S.; Schmidt, M.E.; Baddeley, H.J.E.; Tom, C.M.; Polturi, N.; Xie, Y.; Mattis, V.B.; et al. The Interaction of Aging and Cellular Stress Contributes to Pathogenesis in Mouse and Human Huntington Disease Neurons. Front. Aging Neurosci. 2020, 12, 524369.
- Machiela, E.; Southwell, A.L. Biological Aging and the Cellular Pathogenesis of Huntington’s Disease. J. Huntingt. Dis. 2020, 9, 115–128.
- Pinho, B.R.; Reis, S.D.; Hartley, R.C.; Murphy, M.P.; Oliveira, J.M.A. Mitochondrial Superoxide Generation Induces a Parkinsonian Phenotype in Zebrafish and Huntingtin Aggregation in Human Cells. Free Radic. Biol. Med. 2019, 130, 318–327.
- Liu, L.; Bai, J.; Liu, F.; Xu, Y.; Zhao, M.; Zhao, C.; Zhou, Z. Cross-Talking Pathways of Forkhead Box O1 (FOXO1) Are Involved in the Pathogenesis of Alzheimer’s Disease and Huntington’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 7619255.