Hepatocellular carcinoma (HCC) represents a worldwide health matter with a major care burden, high prevalence, and poor prognosis. Its pathogenesis mainly varies depending on the underlying etiological factors, although it develops from liver cirrhosis in the majority of cases. In the premalignant environment, inflammatory cells release a wide range of cytokines, chemokines, growth factors, prostaglandins, and proangiogenic factors, making the liver environment more suitable for hepatocyte tumor progression that starts from acquired genetic mutations. A complex interaction of pro-inflammatory (IL-6, TNF-α) and anti-inflammatory cytokines (TGF-α and -β), pro-angiogenic molecules (including the Angiopoietins, HGF, PECAM-1, HIF-1α, VEGF), different transcription factors (NF-kB, STAT-3), and their signaling pathways are involved in the development of HCC. Since cytokines are expressed and released during the different stages of HCC progression, their measurement, by different available methods, can provide in-depth information on the identification and management of HCC.
- biomarkers
- cytokines
- hepatocellular carcinoma
- personalized medicine
1. Introduction
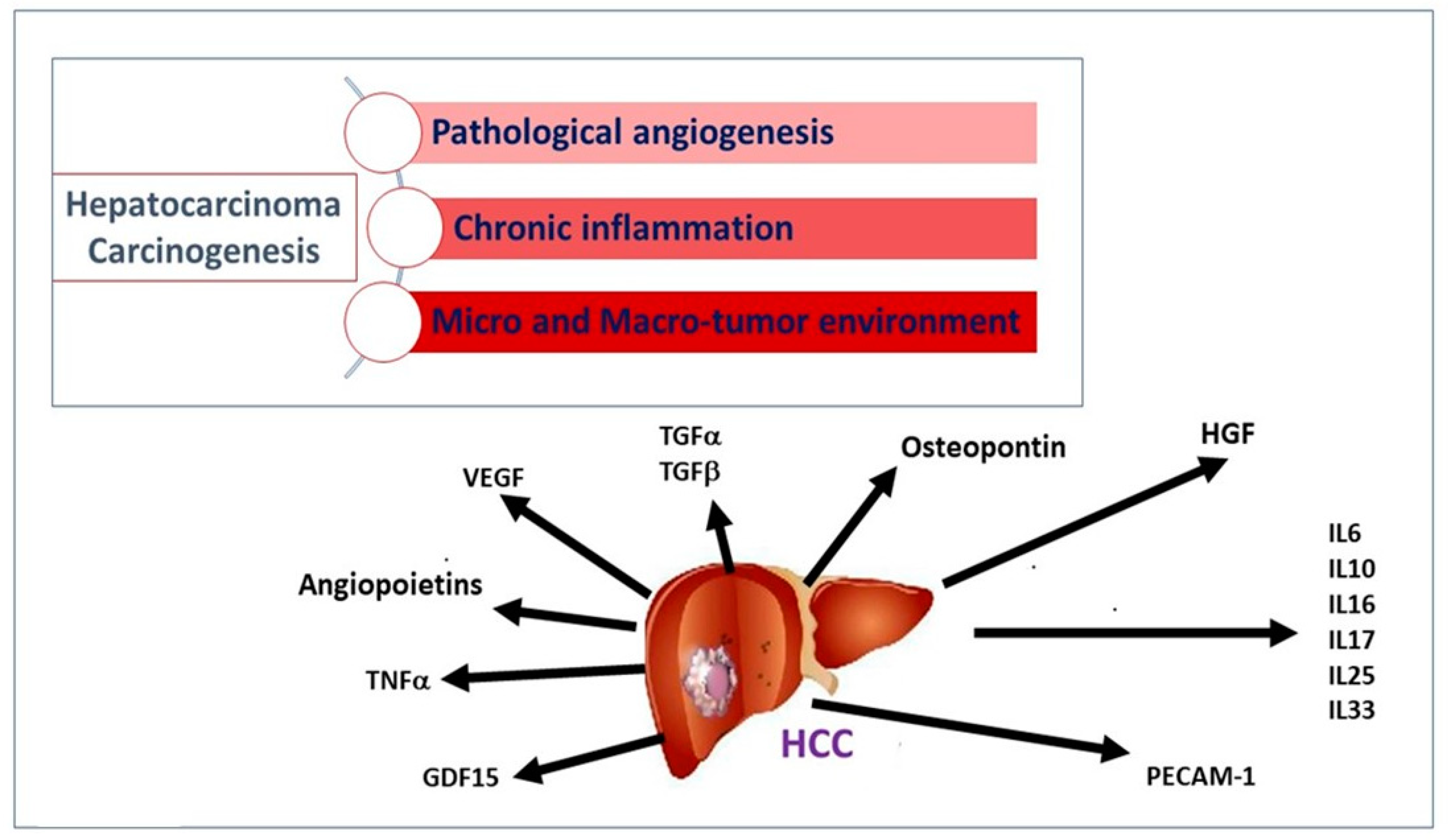
2. Cytokines and Growth Factors
2.1. Stimulators of Angiogenesis and Tumor Invasiveness
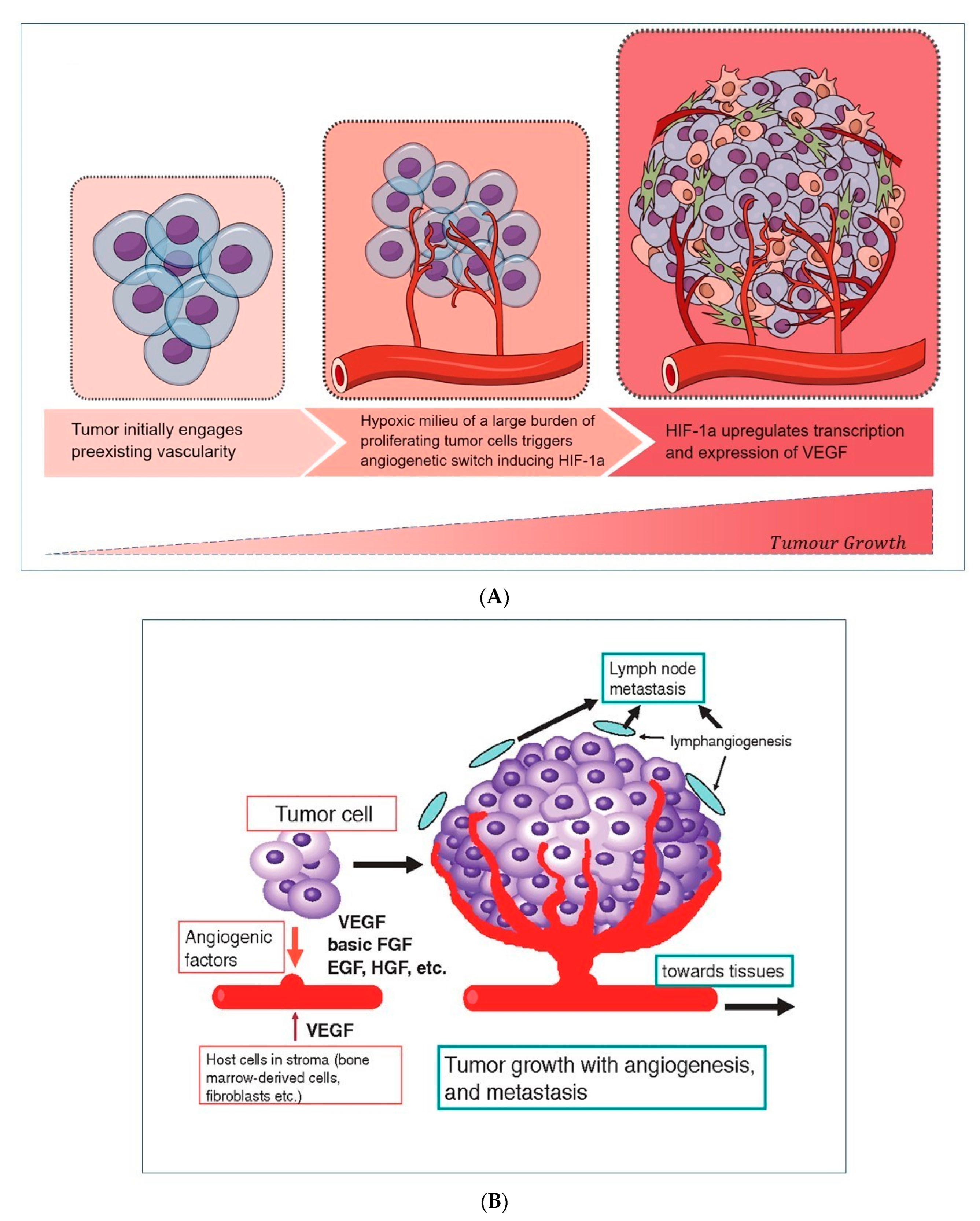
2.2. Stimulators of Chronic Inflammation, Liver Fibrosis, and Proliferation
2.3. Liver Tumor Inducers
3. Detection and Measurement of Cytokines
This entry is adapted from the peer-reviewed paper 10.3390/jpm13010005
References
- AIOM; AIRTUM. I Numeri Del Cancro in Italia, XI Edizione 2021. Available online: https://www.aiom.it/i-numeri-del-cancro-in-italia/ (accessed on 1 January 2021).
- Yang, Y.; Kim, S.; Seki, E. Inflammation and Liver Cancer: Molecular Mechanisms and Therapeutic Targets. Semin. Liver Dis. 2019, 39, 026–042.
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and Chemokines: At the Crossroads of Cell Signalling and Inflammatory Disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582.
- Cabillic, F.; Corlu, A. Regulation of Transdifferentiation and Retrodifferentiation by Inflammatory Cytokines in Hepatocellular Carcinoma. Gastroenterology 2016, 151, 607–615.
- Medina, J.; Arroyo, A.G.; Sánchez-Madrid, F.; Moreno-Otero, R. Angiogenesis in Chronic Inflammatory Liver Disease. Hepatology 2004, 39, 1185–1195.
- Tammela, T.; Enholm, B.; Alitalo, K.; Paavonen, K. The Biology of Vascular Endothelial Growth Factors. Cardiovasc. Res. 2005, 65, 550–563.
- Mukozu, T.; Nagai, H.; Matsui, D.; Kanekawa, T.; Sumino, Y. Serum VEGF as a Tumor Marker in Patients with HCV-Related Liver Cirrhosis and Hepatocellular Carcinoma. Anticancer. Res. 2013, 33, 1013–1021.
- Zekri, A.-R.N.; Bahnassy, A.A.; Alam El-Din, H.M.; Morsy, H.M.; Shaarawy, S.; Moharram, N.Z.; Daoud, S.S. Serum Levels of β-Catenin as a Potential Marker for Genotype 4/Hepatitis C-Associated Hepatocellular Carcinoma. Oncol. Rep. 2011, 26, 825–831.
- Pocino, K.; Napodano, C.; Marino, M.; Di Santo, R.; Miele, L.; De Matthaeis, N.; Gulli, F.; Saporito, R.; Rapaccini, G.L.; Ciasca, G.; et al. A Comparative Study of Serum Angiogenic Biomarkers in Cirrhosis and Hepatocellular Carcinoma. Cancers 2021, 14, 11.
- Dong, G.; Lin, X.-H.; Liu, H.-H.; Gao, D.-M.; Cui, J.-F.; Ren, Z.-G.; Chen, R.-X. Intermittent Hypoxia Alleviates Increased VEGF and Pro-Angiogenic Potential in Liver Cancer Cells. Oncol. Lett. 2019, 18, 1831–1839.
- Miyahara, K.; Nouso, K.; Morimoto, Y.; Takeuchi, Y.; Hagihara, H.; Kuwaki, K.; Onishi, H.; Ikeda, F.; Miyake, Y.; Nakamura, S.; et al. Pro-Angiogenic Cytokines for Prediction of Outcomes in Patients with Advanced Hepatocellular Carcinoma. Br. J. Cancer 2013, 109, 2072–2078.
- Llovet, J.M.; Peña, C.E.A.; Lathia, C.D.; Shan, M.; Meinhardt, G.; Bruix, J. SHARP Investigators Study Group Plasma Biomarkers as Predictors of Outcome in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2012, 18, 2290–2300.
- Gong, L.; Giacomini, M.M.; Giacomini, C.; Maitland, M.L.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Sorafenib Pathways. Pharm. Genom. 2017, 27, 240–246.
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of Vascular Morphogenesis and Homeostasis through the Angiopoietin-Tie System. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177.
- Naldini, A.; Carraro, F. Role of Inflammatory Mediators in Angiogenesis. Curr. Drug Targets. Inflamm. Allergy 2005, 4, 3–8.
- Jeon, B.H.; Khanday, F.; Deshpande, S.; Haile, A.; Ozaki, M.; Irani, K. Tie-Ing the Antiinflammatory Effect of Angiopoietin-1 to Inhibition of NF-KappaB. Circ. Res. 2003, 92, 586–588.
- Sullivan, C.C.; Du, L.; Chu, D.; Cho, A.J.; Kido, M.; Wolf, P.L.; Jamieson, S.W.; Thistlethwaite, P.A. Induction of Pulmonary Hypertension by an Angiopoietin 1/TIE2/Serotonin Pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 12331–12336.
- Koh, G.Y. Orchestral Actions of Angiopoietin-1 in Vascular Regeneration. Trends Mol. Med. 2013, 19, 31–39.
- Fiedler, U.; Augustin, H.G. Angiopoietins: A Link between Angiogenesis and Inflammation. Trends Immunol. 2006, 27, 552–558.
- Fiedler, U.; Reiss, Y.; Scharpfenecker, M.; Grunow, V.; Koidl, S.; Thurston, G.; Gale, N.W.; Witzenrath, M.; Rosseau, S.; Suttorp, N.; et al. Angiopoietin-2 Sensitizes Endothelial Cells to TNF-Alpha and Has a Crucial Role in the Induction of Inflammation. Nat. Med. 2006, 12, 235–239.
- Scharpfenecker, M.; Fiedler, U.; Reiss, Y.; Augustin, H.G. The Tie-2 Ligand Angiopoietin-2 Destabilizes Quiescent Endothelium through an Internal Autocrine Loop Mechanism. J. Cell Sci. 2005, 118, 771–780.
- Sugimachi, K.; Tanaka, S.; Taguchi, K.; Aishima, S.; Shimada, M.; Tsuneyoshi, M. Angiopoietin Switching Regulates Angiogenesis and Progression of Human Hepatocellular Carcinoma. J. Clin. Pathol. 2003, 56, 854–860.
- Scholz, A.; Rehm, V.A.; Rieke, S.; Derkow, K.; Schulz, P.; Neumann, K.; Koch, I.; Pascu, M.; Wiedenmann, B.; Berg, T.; et al. Angiopoietin-2 Serum Levels Are Elevated in Patients with Liver Cirrhosis and Hepatocellular Carcinoma. Am. J. Gastroenterol. 2007, 102, 2471–2481.
- García-Vilas, J.A.; Medina, M.Á. Updates on the hepatocyte growth factor/c-Met axis in hepatocellular carcinoma and its therapeutic implications. World J. Gastroenterol. 2018, 24, 3695–3708.
- Noguchi, O.; Enomoto, N.; Ikeda, T.; Kobayashi, F.; Marumo, F.; Sato, C. Gene Expressions of C-Met and Hepatocyte Growth Factor in Chronic Liver Disease and Hepatocellular Carcinoma. J. Hepatol. 1996, 24, 286–292.
- Ljubimova, J.Y.; Petrovic, L.M.; Wilson, S.E.; Geller, S.A.; Demetriou, A.A. Expression of HGF, Its Receptor c-Met, c-Myc, and Albumin in Cirrhotic and Neoplastic Human Liver Tissue. J. Histochem. Cytochem. 1997, 45, 79–87.
- Vejchapipat, P.; Tangkijvanich, P.; Theamboonlers, A.; Chongsrisawat, V.; Chittmittrapap, S.; Poovorawan, Y. Association between Serum Hepatocyte Growth Factor and Survival in Untreated Hepatocellular Carcinoma. J. Gastroenterol. 2004, 39, 1182–1188.
- Breuhahn, K.; Longerich, T.; Schirmacher, P. Dysregulation of Growth Factor Signaling in Human Hepatocellular Carcinoma. Oncogene 2006, 25, 3787–3800.
- Fodor, D.; Jung, I.; Turdean, S.; Satala, C.; Gurzu, S. Angiogenesis of hepatocellular carcinoma: An immunohistochemistry study. World J. Hepatol. 2019, 11, 294–304.
- DeLisser, H.M.; Christofidou-Solomidou, M.; Strieter, R.M.; Burdick, M.D.; Robinson, C.S.; Wexler, R.S.; Kerr, J.S.; Garlanda, C.; Merwin, J.R.; Madri, J.A.; et al. Involvement of Endothelial PECAM-1/CD31 in Angiogenesis. Am. J. Pathol. 1997, 151, 671–677.
- McCormick, B.A.; Zetter, B.R. Adhesive Interactions in Angiogenesis and Metastasis. Pharmacol. Ther. 1992, 53, 239–260.
- Zhang, Y.-Y.; Kong, L.-Q.; Zhu, X.-D.; Cai, H.; Wang, C.-H.; Shi, W.-K.; Cao, M.-Q.; Li, X.-L.; Li, K.-S.; Zhang, S.-Z.; et al. CD31 Regulates Metastasis by Inducing Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma via the ITGB1-FAK-Akt Signaling Pathway. Cancer Lett. 2018, 429, 29–40.
- Schmidt-Arras, D.; Rose-John, S. IL-6 Pathway in the Liver: From Physiopathology to Therapy. J. Hepatol. 2016, 64, 1403–1415.
- Bartoccioni, E.; Scuderi, F.; Marino, M.; Provenzano, C. IL-6, Monocyte Infiltration and Parenchymal Cells. Trends Immunol. 2003, 24, 299–300, author reply 300–301.
- Marino, M.; Scuderi, F.; Ponte, E.; Maiuri, M.T.; De Cristofaro, R.; Provenzano, C.; Rose-John, S.; Cittadini, A.; Bartoccioni, E. Novel Path to IL-6 Trans-Signaling through Thrombin-Induced Soluble IL-6 Receptor Release by Platelets. J. Biol. Regul. Homeost. Agents 2013, 27, 841–852.
- Kao, J.-T.; Lai, H.-C.; Tsai, S.-M.; Lin, P.-C.; Chuang, P.-H.; Yu, C.-J.; Cheng, K.-S.; Su, W.-P.; Hsu, P.-N.; Peng, C.-Y.; et al. Rather than Interleukin-27, Interleukin-6 Expresses Positive Correlation with Liver Severity in Naïve Hepatitis B Infection Patients. Liver Int. 2012, 32, 928–936.
- Lai, S.-C.; Su, Y.-T.; Chi, C.-C.; Kuo, Y.-C.; Lee, K.-F.; Wu, Y.-C.; Lan, P.-C.; Yang, M.-H.; Chang, T.-S.; Huang, Y.-H. DNMT3b/OCT4 Expression Confers Sorafenib Resistance and Poor Prognosis of Hepatocellular Carcinoma through IL-6/STAT3 Regulation. J. Exp. Clin. Cancer Res. 2019, 38, 474.
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of Liver Cancer Progenitors Whose Malignant Progression Depends on Autocrine IL-6 Signaling. Cell 2013, 155, 384–396.
- Shao, Y.-Y.; Lin, H.; Li, Y.-S.; Lee, Y.-H.; Chen, H.-M.; Cheng, A.-L.; Hsu, C.-H. High Plasma Interleukin-6 Levels Associated with Poor Prognosis of Patients with Advanced Hepatocellular Carcinoma. Jpn. J. Clin. Oncol. 2017, 47, 949–953.
- Zhang, J.; Wang, W.-L.; Li, Q.; Qiao, Q. Expression of Transforming Growth Factor-Alpha and Hepatitis B Surface Antigen in Human Hepatocellular Carcinoma Tissues and Its Significance. World J. Gastroenterol. 2004, 10, 830–833.
- Marino, M.; Scuderi, F.; Mannella, F.; Bartoccioni, E. TGF-Β1 and IL-10 Modulate IL-1β-Induced Membrane and Soluble ICAM-1 in Human Myoblasts. J. Neuroimmunol. 2003, 134, 151–157.
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230.
- Furuta, K.; Misao, S.; Takahashi, K.; Tagaya, T.; Fukuzawa, Y.; Ishikawa, T.; Yoshioka, K.; Kakumu, S. Gene Mutation of Transforming Growth Factor Beta1 Type II Receptor in Hepatocellular Carcinoma. Int. J. Cancer 1999, 81, 851–853.
- Alqahtani, A.; Khan, Z.; Alloghbi, A.; Said Ahmed, T.S.; Ashraf, M.; Hammouda, D.M. Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies. Medicina 2019, 55, E526.
- Gonzalez-Sanchez, E.; Vaquero, J.; Férnandez-Barrena, M.G.; Lasarte, J.J.; Avila, M.A.; Sarobe, P.; Reig, M.; Calvo, M.; Fabregat, I. The TGF-β Pathway: A Pharmacological Target in Hepatocellular Carcinoma? Cancers 2021, 13, 3248.
- Srivastava, A.; Sharma, H.; Khanna, S.; Sadhu Balasundaram, T.; Chowdhury, S.; Chowdhury, R.; Mukherjee, S. Interleukin-6 Induced Proliferation Is Attenuated by Transforming Growth Factor-β-Induced Signaling in Human Hepatocellular Carcinoma Cells. Front. Oncol. 2021, 11, 811941.
- Shakiba, E.; Ramezani, M.; Sadeghi, M. Evaluation of Serum Interleukin-6 Levels in Hepatocellular Carcinoma Patients: A Systematic Review and Meta-Analysis. Clin. Exp. Hepatol. 2018, 4, 182–190.
- Chau, G.Y.; Wu, C.W.; Lui, W.Y.; Chang, T.J.; Kao, H.L.; Wu, L.H.; King, K.L.; Loong, C.C.; Hsia, C.Y.; Chi, C.W. Serum Interleukin-10 but Not Interleukin-6 Is Related to Clinical Outcome in Patients with Resectable Hepatocellular Carcinoma. Ann. Surg. 2000, 231, 552–558.
- Shakiba, E.; Ramezani, M.; Sadeghi, M. Evaluation of serum interleukin-10 levels in hepatocellular carcinoma patients: A systematic review and meta-analysis. Clin. Exp. Hepatol. 2018, 4, 35–40.
- Tiegs, G.; Horst, A.K. TNF in the Liver: Targeting a Central Player in Inflammation. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2022.
- Villanueva, A.; Luedde, T. The Transition from Inflammation to Cancer in the Liver. Clin. Liver Dis. 2016, 8, 89–93.
- Moran, D.M.; Mattocks, M.A.; Cahill, P.A.; Koniaris, L.G.; McKillop, I.H. Interleukin-6 Mediates G(0)/G(1) Growth Arrest in Hepatocellular Carcinoma through a STAT 3-Dependent Pathway. J. Surg. Res. 2008, 147, 23–33.
- Xu, J.; Lin, H.; Wu, G.; Zhu, M.; Li, M. IL-6/STAT3 Is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 760971.
- Cruikshank, W.W.; Kornfeld, H.; Center, D.M. Interleukin-16. J. Leukoc. Biol. 2000, 67, 757–766.
- Takeba, Y.; Ohta, Y.; Ootaki, M.; Kobayashi, T.; Kida, K.; Watanabe, M.; Koizumi, S.; Otsubo, T.; Iiri, T.; Matsumoto, N. Identification of Interleukin-16 Production on Tumor Aggravation in Hepatocellular Carcinoma by a Proteomics Approach. Tumour Biol. 2021, 43, 309–325.
- Brown, L.F.; Berse, B.; Van de Water, L.; Papadopoulos-Sergiou, A.; Perruzzi, C.A.; Manseau, E.J.; Dvorak, H.F.; Senger, D.R. Expression and Distribution of Osteopontin in Human Tissues: Widespread Association with Luminal Epithelial Surfaces. Mol. Biol. Cell 1992, 3, 1169–1180.
- Zhao, H.; Chen, Q.; Alam, A.; Cui, J.; Suen, K.C.; Soo, A.P.; Eguchi, S.; Gu, J.; Ma, D. The Role of Osteopontin in the Progression of Solid Organ Tumour. Cell Death Dis. 2018, 9, 356.
- Da Costa, A.N.; Plymoth, A.; Santos-Silva, D.; Ortiz-Cuaran, S.; Camey, S.; Guilloreau, P.; Sangrajrang, S.; Khuhaprema, T.; Mendy, M.; Lesi, O.A.; et al. Osteopontin and Latent-TGF β Binding-Protein 2 as Potential Diagnostic Markers for HBV-Related Hepatocellular Carcinoma. Int. J. Cancer 2015, 136, 172–181.
- Duarte-Salles, T.; Misra, S.; Stepien, M.; Plymoth, A.; Muller, D.; Overvad, K.; Olsen, A.; Tjønneland, A.; Baglietto, L.; Severi, G.; et al. Circulating Osteopontin and Prediction of Hepatocellular Carcinoma Development in a Large European Population. Cancer Prev. Res. 2016, 9, 758–765.
- Myojin, Y.; Hikita, H.; Tahata, Y.; Doi, A.; Kato, S.; Sasaki, Y.; Shirai, K.; Sakane, S.; Yamada, R.; Kodama, T.; et al. Serum Growth Differentiation Factor 15 Predicts Hepatocellular Carcinoma Occurrence after Hepatitis C Virus Elimination. Aliment. Pharmacol. Ther. 2022, 55, 422–433.
- Kupcova Skalnikova, H.; Cizkova, J.; Cervenka, J.; Vodicka, P. Advances in Proteomic Techniques for Cytokine Analysis: Focus on Melanoma Research. Int. J. Mol. Sci. 2017, 18, E2697.