Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
|
Immunology
Lipid metabolism is the major intracellular mechanism driving a variety of cellular functions such as energy storage, hormone regulation and cell division. Lipids, being a primary component of the cell membrane, play a pivotal role in the survival of macrophages. Lipids are crucial for a variety of macrophage functions including phagocytosis, energy balance and ageing. Lipid-loaded macrophages have recently been emerging as a hallmark for several diseases.
- lipid droplets
- macrophages
- microglia
- autophagy
- hypoxia
1. Introduction
ER remains the key regulator of lipid metabolism in a cell. Around one-third of the proteome is synthesized, matured and modified at the rough ER, bound with ribosomes at the membrane. Biosynthesis of lipids, hormone, steroids and xenobiotic detoxification remains the function of smooth ER [1]. The ER houses the enzymes involved in synthesis of cholesterol and triacylglycerides (TAG) [2]. While TAGs are transferred to lipid droplets (LDs) budding from the ER membrane, lipids synthesized at ER are distributed to other organelles via the secretory pathway. At the ER, the cellular cholesterol is controlled via pathways that sense cholesterol levels within the ER membrane and impart signals to control both synthesis and clearance of cholesterol [3]. Under conditions of low ER cholesterol, the primary regulation of cholesterol at the ER involves synthesis of cholesterol via SCAP/SREBP2 (sterol regulatory-element binding proteins 2) pathway [4] followed by conversion of cholesterol into oxysterols and finally into bile acids [5] and production of cholesterol esters which eventually move into lipid droplets [6]. Similarly, ER regulates the intracellular fatty acid composition to regulate the cellular demands required for synthesizing complex lipids. Moreover, ER remains the key regulator of fatty acid synthesis and lipid metabolism. Structural modification of fatty acids such as elongases, desaturases and beta-oxidation cycles occur at ER [7]. SREBPs, a family of membrane bound transcription factors are actively involved in lipid homeostasis. SREBPs, synthesized as precursors reside at the ER membrane [8]. While SREBP1a functions mainly in lipid synthesis in proliferating cells, the major role of SREBP1c remains in regulating the synthesis of triglycerides (TG) and fatty acids in lipogenic organs. SREBP2 widely regulates synthesis of sterols in tissues [9,10]. De novo synthesis of fatty acids is also partially regulated at the ER through feedback inhibition mechanisms of SREBP-1 release [11]. Further, under stimulated TAG synthesis condition, enzymes involved in the biosynthesis of TAG (lipin, DGAT, GPAT and AGPAT) relocalize from ER to lipid droplets [12].
Additionally, synthesis of lipids occurs also at the ER-organelle contact sites. Lipids are trafficked out of ER by specialized ER domains via lipid transfer proteins [2]. It is scientifically plausible that lipid droplets are formed in ER at the regions where synthesis of triacylglycerols (TAGs) or sterol esters takes place [13]. Phospholipids and proteins are also biochemically modified at the ER–Golgi intermediate compartment (ERGIC) and are distributed within the cell via secretory pathways or direct organelle contacts [14,15]. Phospholipids and neutral lipids (TG and CE) are the two primary forms of lipids the ER is comprised of. The key function of phospholipids includes the assembly of membranes and vesicles involved in protein trafficking; TG and CE function as reserves for excess cholesterol and fatty acids and owing to their hydrophobicity, they instigate formation of LDs within the ER membrane [16]. Interestingly, ER -resident DGAT2 (diglyceride acyltransferase) enzyme, pivotal for the synthesis of TG, mediates synthesis and storage of TG in lipid droplets independent of its localization in ER [17]. Further, structurally uniform ER–LD contacts along with the delivery of TGs from ER to LDs were reported to be facilitated by an ER integral protein called seipin [18].
It is also notable that the PERK-eIF2α pathway regulates lipogenesis. For example, antipsychotic drugs induced phosphorylation of PERK and eIF2α, resulting in SREBP-1c and SREBP2 mediated accumulation of lipids in hepatic cells [27] (Figure 1).
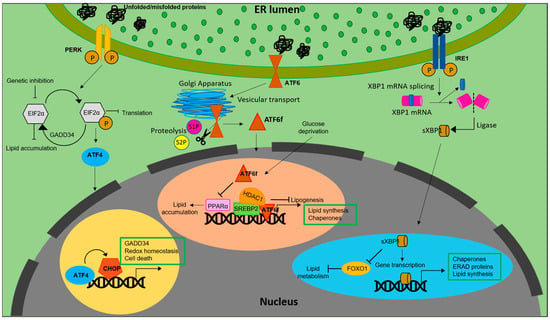
Figure 1. Lipid biosynthesis at ER. The schematic representation shows the mechanism of lipid biosynthesis taking place at the ER via the three branches of the UPR: ATF6, PERK and IRE1.
Similarly, inhibiting eIF2α phosphorylation via overexpression of growth arrest and DNA damage-inducible gene 34 (GADD34) in liver decreased the hepatosteatosis in mice fed with a high fat diet (HFD) [28]. Genetic deletion of eIF2α aggravated tunicamycin-induced accumulation of lipids in liver [29]. ER stress inducers, brefeldin A and tunicamycin, were reported to induce LD accumulation in Saccharomyces cerevisiae [30]. Further, ATF4, present downstream of the PERK-eIF2α pathway, plays a pivotal role in regulation of lipid metabolism. High carbohydrate diet fed Atf−/− mice displayed less accumulation of TG in liver compared to wild type [31]. Correspondingly, mice lacking ATF4 showed diminished lipid accumulation under conditions of high fructose diet due to decreased levels of FAS, acetyl CoA carboxylase (ACC) and SREBP-1c [32]. White adipose tissue of Atf deficient mice displayed increased lipolysis and decreased lipogenic genes, indicating a direct link between ATF4 and lipid metabolism [33]. Similarly, overexpressing ATF4 instigated early onset of dyslipidemia in zebrafish [34]. The involvement of (C/EBP) homologous protein (CHOP), a protein downstream of the UPR, in the regulation of lipid metabolism emanates from its role in suppressing the gene expression of SREBPF1, CEBPA, and PPARα-like master regulators of lipid metabolism [35]. Following ER stress, CHOP was reported to be critical for the regulation of cholesterol catabolism in macrophages and is involved in the lipid metabolism disorder mediated by ER stress [19]. In mammary epithelium and mouse embryonic fibroblasts that differentiate into adipocytes, absence of PERK resulted in the attenuation of lipogenesis and expression of genes including SREBP1. This led to a decrease in the level of TG and FA content in mammary glands and growth retardation in pups. The study also demonstrated the role of PERK and eIF2α in insig1 translation responsible for SREBP1 activation [36].
Another transmembrane signal transducer of the UPR, the inositol requiring enzyme 1 (IRE1α), is an ER stress sensor, conserved from yeast to mammals [37,38]. During ER stress, IRE1α is activated and splices X-box-binding protein 1 (XBP1) mRNA to its active spliced form to regulate expression of genes involved in restoration of ER homeostasis and biogenesis [39]. In addition to promoting cell survival through attenuation of ER stress, IRE1α also functions as a nutritional stress sensor [40]. Further, IRE1α was found to regulate lipid secretion and lipogenesis in both XBP1-dependent and an independent manner [41,42] (Figure 1). In c-Myc-overexpressing and IRE1α inhibited BL cells, defects in growth and viability were triggered due to altered lipid homeostasis [43]. The IRE1a/XBP1 signaling pathway transcriptionally regulates genes that are players of lipid metabolism in order to activate hepatic lipid metabolism. IRE1α siRNA increased TG and cholesterol levels in XBP1-deficient mice. This suggests that IRE1α hyperactivation reduced plasma lipids under XBP1-deficient conditions. Ablation of XBP1 decreased hepatoxicity [44]. However, how IRE1α functions to maintain lipid homeostasis in peripheral adipose tissues across species remains an enigma. As reviewed by Basseri et al., accumulating evidences suggest that IRE1α is the key component involved in the suppression of hepatic lipid accumulation under conditions of severe ER stress [45]. Under ER stress, IRE1α plays a pivotal role in hepatocytic secretion of LDL and VLDL [42]. XBP1s reduces lipid accumulation by promoting protein degradation of Forkhead box protein O1 (FOXO1) in cardiomyocytes. Similarly, overexpressing XBP1s specifically in cardiomyocytes, mitigated cardiac steatosis [46].
In contrast to IRE1α and PERK, activation of ATF6 does not involve phosphorylation. Under conditions of ER stress, ATF6 is released from BiP, after which the Golgi-localization sequences on the luminal domain of ATF6 are exposed [47] Once transported to Golgi, ATF6 undergoes site-1 (S1P) and site-2 (S2P) proteases mediated cleavage, releasing a cytosolic fragment called ATF6f containing a basic leucine zipper (bZIP) transcription factor [22,48]. The transcriptionally active ATF6 fragment enters the nucleus to trigger a set of transcriptional signaling, to reestablish the ER homeostasis [49,50,51]. Unlike PERK and IRE1 branches of UPR, ATF6 does not function to decrease the flux of unfolded proteins into the ER. Instead, the active fragment induces expansion of the ER membrane in an XBP1-independent manner [52] It also upregulates ER chaperones, ERAD components and disulfide oxidoreductases of the ER lumen [53]. However, ATF6 and XBP1 generated from the IRE1 branch of UPR tend to act synergistically and heterodimerize [54]. ATF6 activation downregulates PPARα, leading to accumulation of lipid droplets resulting in cell death. In contrast, deficiency of ATF6 increased PPARα levels and decreased lipid accumulation and cell death [55]. ATF6 was reported to modulate SREBP2 mediated lipogenesis. Under glucose-deprived conditions, ATF6 interacts with the processed form of SREBP2 to inhibit cholesterol synthesis promoting recruitment of HDAC1. This inhibits the SREBP2-induced lipogenesis and downregulates LDLR expression in HepG2 cells [56].
2. Functions of Lipids in Macrophages of Different Tissue Location
2.1. Lipids and Microglia
Microglia are specialized immune cells resident in the central nervous system (CNS) of the brain and play a key role in the maintenance of brain homeostasis [59,60,61]. Originating from the yolk sac, they migrate to the CNS during embryogenesis; there they propagate and disperse in the CNS in a non-heterogenous manner [62,63]. In a healthy resting brain, microglia are reported to be dynamic and constantly moving [64]. They critically survey the brain environment and get activated upon changes in the brain microenvironment [65]. Their functions include phagocytosis of apoptotic bodies and debris, neuronal protection [64], synaptic remodeling [66,67], neuronal support [68,69] and oligodendrogenesis [70,71]. Microglia dysfunction is a salient feature in neurodegenerative and neuroinflammatory diseases. They become dysfunctional with aging; dysfunctionalities include poor cholesterol efflux, impaired phagocytosis and increased secretion of cytokines and accumulation of lipid droplets [59,72]. Dysregulated lipid metabolism is a characteristic feature observed in neurodegenerative diseases, notably, in Parkinson’s disease (PD) and Alzheimer’s disease (AD). Lipid metabolism in microglia is tightly regulated both during development and disease. Microglia play an inevitable role in maintaining the myelin dynamics. During early development, microglia phagocytose myelin debris and apoptotic oligodendrocytes. The myelin-derived lipids are then cleared by microglia for remyelination post demyelination. Such active clearance and ability to effectively efflux cholesterol is impaired in aging microglia [73,74]. This leads to accumulation and crystallization of cholesterol-rich myelin debris, resulting in defective phagocytosis. Microglia with defective phagocytosis tend to produce large amounts of pro-inflammatory cytokines and reactive oxygen species (ROS) leading to the progression of neuroinflammatory and neurodegenerative diseases [75,76,77]. In a recent study, Loving et al., elucidated the role of lipoprotein lipase (LPL) in the accumulation of LDs and transcriptional regulation of lipid metabolism in in vitro and ex vivo systems. They reported that LPL regulates lipid metabolism in microglia and that loss of LPL resulted in microglial cholesterol load [72]. Phagocytosis deficit can be the consequence of lipid droplets accumulation. Active degradation of lipid droplets and release of free fatty acids was associated with effective phagocytosis [78].
Similarly, lipid droplet accumulating macrophages (LAMs) show downregulated expression of two key enzymes ADRB1 and ADRB2, involved in lipid degradation [59]. Additionally, lipid droplet accumulation promotes transcriptional modulation, giving LAMs a unique transcriptomic signature. Further, Patel and Tulsi et al. reported the association of pathways of lipid and carbohydrate metabolism with sex, age and ApoE expression in human microglia [79]. ApoE is a protein that mediates metabolism and transportation of cholesterol and is prominently expressed in disease-associated microglia (DAM). Moreover, it was reported that extracellular ApoE can be a ligand of TREM2 (triggering receptor expressed on myeloid cells 2) and that ApoE expression is TREM2-dependent [80,81]. Analysis of cell-specific lipidomics reveal that TREM2 deficiency mediates dysregulation of genes associated with lipid metabolism and leads to cholesterol ester overload in microglia [82]. Understanding the link between lipids and neuropathology of NPC (Niemann–Pick disease) patients revealed that loss of NPC1, an intracellular cholesterol transporter in microglia resulted in enhanced uptake of myelin but impaired myelin turnover. Macrophages derived from the blood of NPC patients were found to be similar to the pathological alterations exhibited by microglia of Npc1−/− mice. It was also revealed that Npc1−/− deficient microglia accumulated undigested lipid materials, indicating the role of Npc1 in lipid trafficking in microglia [83].
2.2. Lipids in Adipose Tissue Macrophages (ATMs)
Initially discovered for their role in microbial killing and phagocytosis, macrophages are now known to have distinct and context-dependent functions in different physiological settings. Lipids are the major source of energy for macrophages. Cell membranes of macrophages and precursors of bioactive lipids are provided by lipids. Lipids are also known to regulate the signal transduction during macrophage activation. Activation or polarization of macrophages are dependent on environmental stimuli, which are even tissue-specific, that dictate them to take up unique functions.
Adipose tissue macrophages (ATMs) are key players in metabolic diseases and obesity-associated inflammation. Circulating monocytes accumulating in adipose tissue lead to the development of ATMs [84]. In a study conducted by Prieur et al. in obese mice, it was observed that increase in the accumulation of lipids in ATMs resulted in polarization of macrophages into M1 phenotype, a phenotype associated with insulin resistance and obesity. Their results indicate that M1 polarization of ATMs are associated with accumulation and proliferation of lipid species, giving them the resemblance of vascular foam cells (discussed later). In addition to M1 polarization, ATMs of obese mice strongly accumulated lipids in their cytoplasm, resembling pro-atherosclerotic vascular foam cells. Increased fat deposition in adipose tissue decreased the expandability of adipose tissue, leading to adipocyte dysfunction and lipid leakage and thereafter lipid accumulation in ATMs [85]. Similarly, M1 macrophages treated with exogenous fatty acids showed an increase in TG and CE levels. The accumulation of exogenous fatty acids was high in M2 macrophages, revealing the impact of macrophage polarization on lipid composition and endogenous lipid pools [86]. ATMs of obese individuals crucially function to scavenge and eliminate adipocyte debris. Under increasing conditions of adiposity, excess lipid species are stored in ATMs, leading to the formation of lipid-laden ATM population [87]. Cd36, Fabp4, Fabp5 and Lpl are the set of genes that are highly conserved in lipid-associated ATMs [88].
Lipid-associated macrophages (LAMs) are distinctly conserved subset of macrophages predominantly expanded in adipose tissues of obese individuals. The formation of LAMs in adipose tissues are driven by a TF called TREM2. TREM2-regulated TAMs were reported to be inevitable during the loss of adipose tissue homeostasis, as they prevent metabolic disorders [88]. Suppression of tumor growth of triple-negative breast cancer (TNBC) was achieved by genetically depleting LAM subsets [89].
Atherosclerosis is another condition where the lipid homeostasis of macrophages is disrupted. During atherosclerosis, initially, the modified lipoproteins and serum lipids are deposited under the endothelial cells, activating them to secrete adhesion molecules. Circulating monocytes interact with these adhesion molecules that adhere to the endothelial cells and migrate to subendothelial space where they are differentiated into macrophages. Ingestion of modified lipoproteins by macrophages takes place via receptor mediated phagocytosis or pinocytosis. As a result of excess uptake of lipids, macrophages tend to store the excess neutral lipids in the form of lipid droplets in the cytoplasm. Excess accumulation of lipid droplets (LDs) in macrophages gives them a foamy appearance and hence they are called “macrophage foam cells”. These lipid-laden macrophages are the hallmark of atherosclerosis.
Formation of lipid-laden macrophage foam cells in lungs occur even during the infection of Mycobacterium tuberculosis. In response to TB infection, macrophages undergo metabolic changes and develop into foam cells. Though they resemble atherosclerotic foam cells, their lipid composition and roles remain different. Unlike atherosclerotic foam cells, their lipid content is predominantly triglycerides (TG) and not cholesterol. Here, the formation of TB foam cells is attributed to mycolic acid from pathogens. These TB foam cells often dominate the mycobacterial granulomata associated with caseum [90].
2.3. Lipids in Tumor Associated Macrophages (TAMs)
TAMs are macrophages present in the microenvironment of solid tumors, creating an immunosuppressive environment. Lipids play a key role in the development of TAMs in the tumor microenvironment (TME). Accumulating evidence suggest that abnormal lipid accumulation is inevitable for TAMs to engage in protumorigenic activity. Tumorigenesis and tumor progression is associated with the functional plasticity of TAMs which is often dictated by their metabolic features. The metabolic and functional landscape of tumor cells keeps evolving according to the selective pressure of the inconsistency in the availability of nutrients and oxygen in the TME, as a result of which functional features of TAMs are often altered [91]. Macrophages from both murine and human tumors were found to express high levels of a scavenger receptor, CD36, and ingest more lipids [92] for use as source of energy via oxidative phosphorylation and fatty acid oxidation. Contradictorily, enhanced fatty acid oxidation in macrophages caused by fatty acids in tumor microenvironment results in increased ROS production and decreased IL-10 secretion to eliminate tumor cells. This signifies the involvement of lipid metabolism in anti-tumor response [93]. Similarly, in prostatic adenocarcinoma (PCa), TAMs in the TME were shown to have dysregulated lipid metabolism. Accumulation of lipids in TAMs was reported to positively correlate with the progression of PCa [94]. TAMs characterized as M2-like cells, suppress tumor immune surveillance to promote tumor growth and metastasis. In a study, Wu et.al., demonstrated that enhancing lipid metabolism is sufficient for modulating the phenotype of macrophages into immunosuppressive TAMs [95]. Further, ingestion of a high amount of lipids from tumor cells leads to over expression of phosphoinositide 3-kinase (PI3K-γ) resulting in the polarization of TAMs into an M2-like phenotype. Inhibiting PI3K-γ reversed the pro-tumor phenotype of LD-loaded TAMs, suppressing the growth of gastric cancer [96]. In a study conducted on a melanoma model, it was reported that β-glucosylceramide released by tumor cells served as a stimulus for protumorigenic polarization of TAMs via induction of ER stress responses-mediated shuffling of lipid composition in macrophages [97]. In addition to lipid accumulation, TAMs exhibit decreased phagocytic activity with upregulated expression of programmed death ligand 1 (PD-L1); they block anti–tumor T cell responses to support immunosuppression [96]. From these reports, it is scientifically evident that, lipid droplets are critical cell structures that can be targeted for the development of a novel anti-tumor strategy and that reprogramming lipid metabolism can maximize the impact of anti-tumor therapies.
2.4. Lipids in Phagocytic Function of Macrophages
Macrophages are phagocytic in nature. Through phagocytosis, macrophages engulf foreign organisms and other invading pathogens, thereby defending the host against infection. The crosstalk between hypoxia and inflammation has a significant implication for infection and sterile inflammation in macrophages. Macrophages are the primary component of the innate immune response that is known for phagocytosis of invading pathogens and microorganism. Apoptotic cells are also eliminated by macrophages via phagocytosis. Formation of lipid-rich organelles, called lipid bodies or lipid droplets (LDs), occur in parallel with formation and maturation of phagosomes containing pathogens [98,99]. Infections with microbes such as bacteria, virus and other parasites induced LD accumulation in immune cells both clinically and experimentally [100,101]. These lipid bodies, formed in response to infections relocate within cytoplasm to interact with the phagosomes [102]. However, this association between LDs and phagosomes is yet ill-understood. Nevertheless, this interaction is accounted for the survival of pathogens within host cells.
2.5. Oxidized Phosphocholines in the Immune Function of Macrophages
Phosphocholines, belonging to the class of phospholipids, are a vital component of mammalian cells. Cell death occurring in the local environment of inflammatory or non-inflammatory tissue injury results in ROS generation. This can oxidize the phosphocholines present in the plasma membrane. Upon exposure to ROS, arachidonic acid-containing phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC), the membrane component of mammalian cells, is oxidized at different positions, creating a heterogenous mixture of lipids called oxPAPCs. The scavenger receptor CD36 present on macrophages, induce the uptake of oxPAPCs. This leads to formation of foam cells during atherosclerosis [103]. The internalization of oxPAPCs into endosomes and their transport to cytosol is also mediated by bacterial lipopolysaccharide (LPS) receptor, CD14 present on myeloid cells [104]. Post recognition by macrophages, oxPAPCs exert proinflammatory or anti-inflammatory activities based on the context in which they were encountered [105]. Pre-treatment with oxPAPCs modulated the phagocytosis of bacteria in macrophages [106]. In contrast, oxPLs trigger CD36-mediated phagocytosis of apoptotic cells in macrophages [107]. Further, oxPAPCs perform a metabolic rewiring in macrophages which is speculated to increase mitochondrial fitness. This, in turn, could prolong their lifespan [108]. In addition, pure lipids present in oxPAPCs contribute to metabolic hyperinflammation. In addition, oxPAPCs are stimulators of inflammation in airways. During lung injury, oxPAPCs promote secretion of IL-6 from alveolar macrophages [109]. In contrast, pre-treatment of macrophages with oxPAPCs was reported to block the response of nuclear factor-κB to LPS treatments. The competitive interaction of LPS and oxPAPCs with CD14 remains the underlying mechanism [110]. The mechanism of oxPAPCs mediated accumulation of lipids in macrophages is shown in Figure 2.
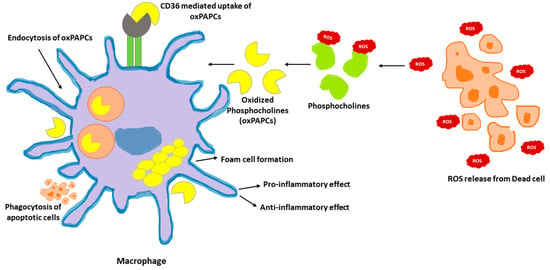
Figure 2. oxPAPCs in the regulation of macrophage immune function. Oxidized by the reactive oxygen speices (ROS) released from dead cells, phosphocholines are taken up by macrophages via endocytosis or CD36 scavenging receptors. Internalized oxPAPCs lead to the formation of foam cells by causing lipid accumulation in macrophages. The oxPAPCs accumulated macrophages are both pro- and anti-inflammatory based on the context.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24010589
This entry is offline, you can click here to edit this entry!