Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The development of diversity-oriented synthesis based on fluorine-containing building blocks has been one of the hot research fields in fluorine chemistry. β-CF3-1,3-enynes, as one type of fluorine-containing building blocks, have attracted more attention due to their distinct reactivity. Numerous value-added trifluoromethylated or non-fluorinated compounds which have biologically relevant structural motifs, such as O-, N-, and S-heterocycles, carboncycles, fused polycycles, and multifunctionalized allenes were synthesized from these fluorine-containing building blocks.
- organofluorine compounds
- fluorine-containing building blocks
- β-CF3-1,3-enynes
1. Introduction
Increasing demands for organofluorine compounds in human life [1][2][3][4][5][6][7][8][9][10][11][12] prompt chemists to develop many ingenious strategies to introduce the fluorine element into organic compounds [13][14][15][16][17][18][19][20][21][22][23][24][25][26][27]. Among these strategies, synthesis with fluorine containing building blocks is a very important method for the introduction of fluorine atoms or fluoroalkyl groups into target molecules [28][29][30][31][32][33][34][35][36]. β-CF3-1,3-enynes as one type of fluorine-containing building blocks have attracted more attention in the last few years due to their distinct reactivity. Numerous value-added fluorine-containing compounds which have biologically relevant structural motifs, such as O-, N-, and S-heterocycles, carboncycles, fused polycycles, and multifunctionalized allenes, were synthesized from these fluorine-containing building blocks.
2. Construction of Trifluoromethylated Carboncycles
2.1. Construction of Three-Membered Trifluoromethylated Carboncycles
In 2019, Wang and Liu reported highly diastereoselective cyclopropanation reactions of β-CF3-1,3-enynes with sulfur ylides via a maneuverable one-pot, two-step procedure. β-CF3-1,3-enynes undergo cyclopropanation reactions with sulfur ylides under mild reaction conditions without fluoride elimination, which affords the cis-isomer mainly. Interestingly, a sequential TBAF-mediated deprotection of the triisopropylsilyl group results in a diastereoenriched epimerization which gives rise to the transcyclopropanes as the sole isomers (Scheme 1a) [37]. A base-triggered thermodynamic epimerization took place during the process, resulting in stereoselectivity enrichment (Scheme 1b). This newly developed protocol was then applied to 2-CF3-3,5-diyne-1-enes for synthesis of diverse 1,3-diyne-tethered cyclopropanes [38].
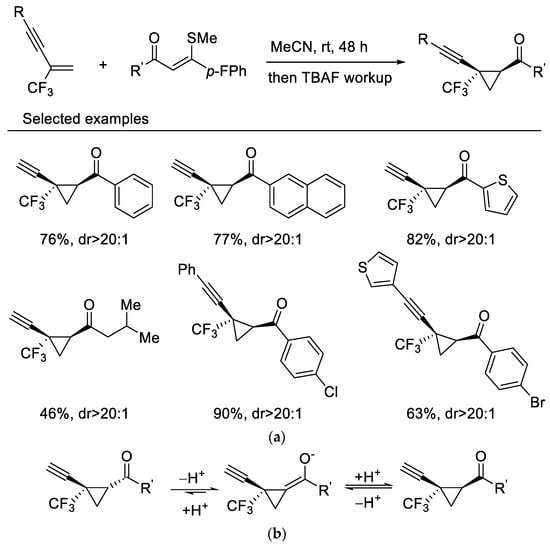
Scheme 1. (a) Synthesis of trifluoromethylated cyclopropanes. (b) The mechanism of thermodynamic epimerization.
Recently, a highly efficient solvent-controlled synthesis of bis(trifluoromethyl) cyclopropanes and bis(trifluoromethyl)pyrazolines via a [2 + 1] or [3 + 2] cycloaddition reaction of β-CF3-1,3-enynes with CF3CHN2 was developed by Cao and co-workers. The distribution of cyclopropanes and pyrazolines is remarkably dependent on the polarity of the solvent used. Less polar solvents, such as DCE, were suitable for the [2 + 1] cycloaddition reaction, whereas polar solvents, such as DMAc, were found to favor the [3 + 2] cycloaddition reaction [39] (Scheme 2).
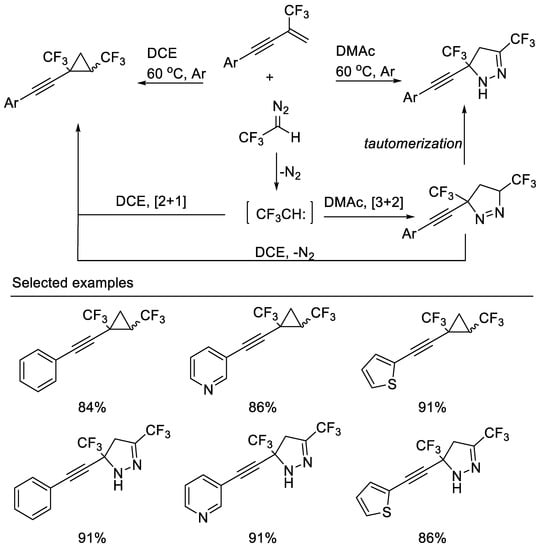
Scheme 2. Divergent synthesis of bis(trifluoromethyl) cyclopropanes and pyrazolines.
2.2. Construction of Five-Membered Trifluoromethylated Carboncycles
In 2011, Jeong and coworkers reported Pd-catalyzed intramolecular carbocyclization of 2-trifluoromethyl-1,1-diphenyl-1,3-enynes to afford 2-trifluoromethyl-1-methylene-3-phenylindene derivatives via electrophilic hydroarylation by the use of 10 mol% Pd(OAc)2 in the presence of CF3CO2H and CH2Cl2. It was postulated that this reaction proceeds via ortho-palladation of enynes to give a corresponding intermediate, which undergoes the insertion to a triple bond to give the vinylpalladium species. Protiodepalladation of vinylpalladium species affords 1-methylene indenes. The substrates were synthesized from pentafluoroethyl phenyl dithioketal in several steps. Both aryl and alkyl-substituted 1,3-enynes are tolerated, however, the stereoisomer ratios of 5/4 of 1-methylene indenes were afforded in this 5-exo-dig carbocyclization. In the case of carbocyclization of trimethylsilyl- or triisopropylsilyl-substituted 1,3-enynes, reduced product was obtained [40] (Scheme 3).
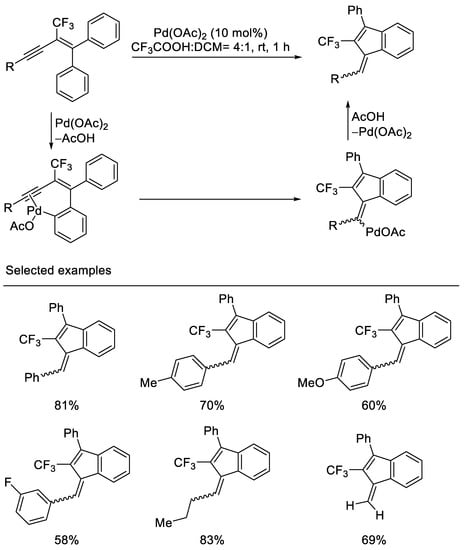
Scheme 3. Synthesis of trifluoromethylated 1-methylene indenes derivatives.
In an elegant piece of work, The Trost group reported palladium-catalyzed trimethylenemethane cycloadditions with α-trifluoromethyl-styrenes, trifluoromethyl-enynes, and dienes under mild reaction conditions. The trifluoromethyl group serves as a unique σ-electron-withdrawing group for the activation of the olefin toward the cycloaddition. This method allows for the formation of exomethylene cyclopentanes bearing a quaternary center substituted by the trifluoromethyl group (Scheme 4) [41]. Diaminophosphite ligand was employed in this reaction to afford desired cycloaddition product in moderated to excellent yields (45–93%). The obtention of the cycloadduct unaccompanied by fluoride elimination may be suggestive of a concerted mechanism.
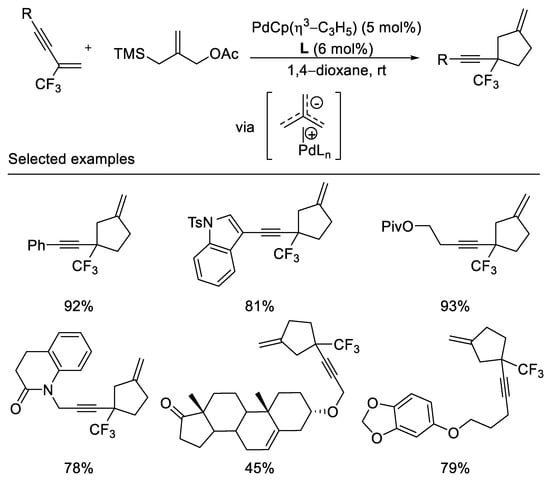
Scheme 4. Synthesis of trifluoromethylated exomethylene cyclopentanes.
In 2017, researchers developed a concise approach to access ring-trifluoromethylated cyclopentene frameworks, utilizing silver-catalyzed double hydrocarbonation reaction of β-CF3-1,3-enynes with bisnucleophiles 1,3-dicarbonyl compounds [42] (Scheme 5). β-CF3-1,3-enynes possessing electron-withdrawing aryl groups on the alkyne moiety smoothly underwent the cyclization reaction with 1,3-dicarbonyl compounds to give ring-trifluoromethylated cyclopentene in moderate to excellent yields. A series of β-diketones, malonate derivatives as well as β-ketoesters could be employed as reaction partner to afford the corresponding ring-trifluoromethylated cyclopentenes in good to excellent yields. Unsymmetrical 1,3-diketone or β-keto esters afford the desired cyclopentenes as a mixture of two diastereomers in excellent yields with moderate to good diastereoselectivity. The use of organic base effectively suppressed the defluorination process.
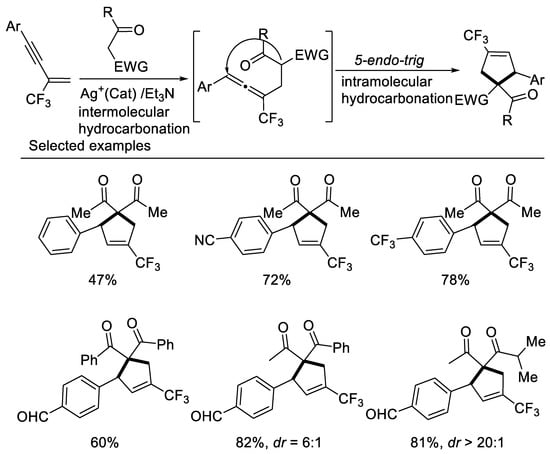
Scheme 5. Synthesis of ring-trifluoromethylated cyclopentene derivatives.
Interestingly, Chang and Zhang recently revealed that inorganic base K3PO4 can effectively promote synthesis of CF3-substituted cyclopentenes when using malononitrile as carbon nucleophile to react with β-CF3-1,3-enynes. Their developed protocol disclosed that the fluorine retention of β-CF3-1,3-enynes did not rely on organic base and the additional patterns did not depend on electron-deficient β-CF3-1,3-enynes. The reasons may mainly be due to the stability of in situ formed carbon anion intermediates containing the cyano group and CF3-substituted cyclopentenes with double bond migration formed [43] (Scheme 6).
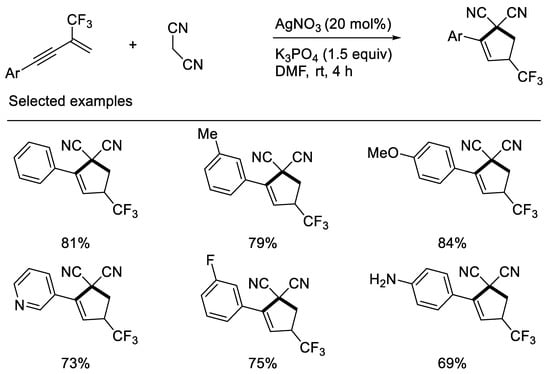
Scheme 6. Synthesis of CF3-substituted cyclopentene derivatives.
In 2020, Liu’s group reported the phosphine-catalyzed [3 + 2] cycloadditions of trifluoromethyl enynes/enediynes with allenoates to form cyclopentenes containing a CF3-substituted quaternary carbon center with great regioselectivity [44] (Scheme 7). This reaction occurs with excellent regioselectivity under mild conditions, affording alkyne- and diyne-tethered cyclopentene derivatives containing a CF3-substituted quaternary carbon center in moderate to good yields.
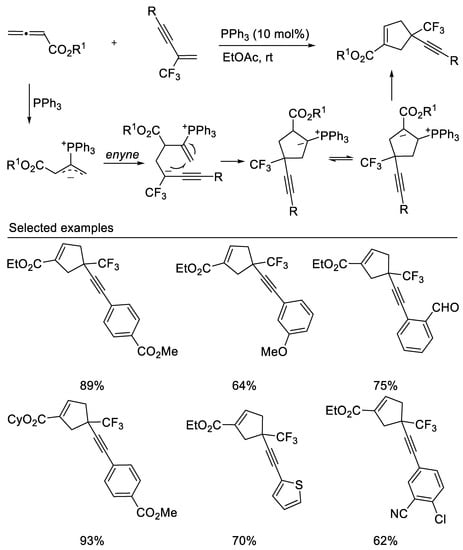
Scheme 7. Phosphine-catalyzed [3 + 2] cycloadditions.
2.3. Construction of Six-Membered Trifluoromethylated Carboncycles
In 2013, the Gevorgyan group demonstrated the synthesis of trifluoromethylated benzene derivatives via chemo- and regioselective Pd-catalyzed [4 + 2] cross-benzannulation of β-CF3-1,3-enynes with diynes [45] (Scheme 8). Both β-F-1,3-enynes and β-perfluoroalkylated 1,3-enynes also are good reaction partners in these Pd-catalyzed [4 + 2] cross-benzannulation reaction affording corresponding trifluoromethylated and perfluoroalkylated benzene derivatives. This cycloaddition strategy proved to be effective for the rapid construction of aromatic fluorides from easily available acyclic starting materials.
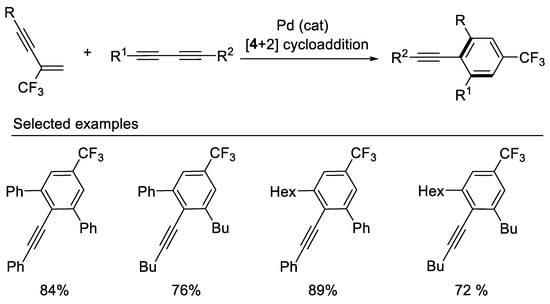
Scheme 8. Synthesis of trifluoromethylated benzene derivatives.
In 2014, the Zhang group disclosed an iron-promoted electrophilic annulation of trifluoromethyl-containing aryl enynes with disulfides or diselenides affording polysubstituted naphthalenes in moderate to excellent yields. The reaction proceeded with high selectivity to provide the 6-endo-dig cyclization product and showed good functional group tolerance. The authors observed that the aryl groups bearing electron-withdrawing substituents in these substrates results in lower yields. The utilization of inexpensive ferric chloride and commercially available disulfides and diselenides as electrophiles are significant advantages for the usefulness of this reaction [46] (Scheme 9).
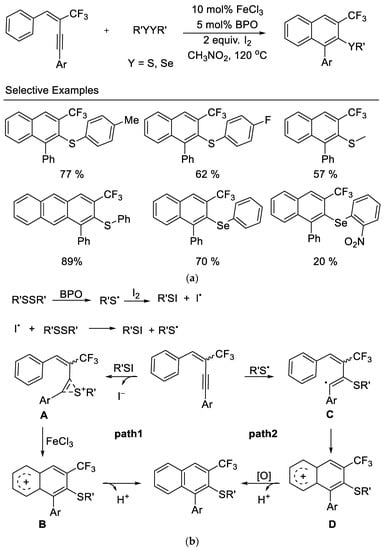
Scheme 9. (a) Synthesis of trifluoromethylated naphthalenes derivatives. (b) Possible mechanism for synthesis of trifluoromethylated naphthalenes derivatives.
Based on their experimental results, a plausible mechanism was outlined, as shown in Scheme 9b. The reaction of disulfide with iodine may take place to produce the active RSI in situ. The presence of BPO promotes the generation of free radical RS• which subsequently reacted with I2 to form RSI. The electrophilic addition of RSI to the triple bond of enyne affords intermediate A (Path 1). The Lewis acid (FeCl3)-catalyzed intramolecular electrophilic attack on the neighboring aryl group provides intermediate B, and subsequent deprotonation yields the desired products. However, a free radical pathway cannot be ruled out (Path 2), in view of the fact that the product can be isolated in the absence of I2. The addition of RS• to enyne produces a vinyl radical C, which then undergoes an intramolecular cyclization to form radical D. The following oxidation by I2 or FeCl3 produces the corresponding carbocation, which loses a H+ to yield desired products.
3. Construction of Trifluoromethylated Heterocycles
3.1. Construction of Five-Membered Trifluoromethylated Heterocycles
Recently, Hanamoto and co-workers developed a convenient method for the synthesis of 4-CF3-3-iodo-2-substituted thiophene from (Z)-2-bromo-2-CF3-vinyl benzyl sulfide in two steps. The Sonogashira cross-coupling reaction of (Z)-2-bromo-2-CF3-vinyl benzyl sulfide with various terminal acetylenes afforded the corresponding (E)-2-CF3-1-buten-3-ynyl benzyl sulfides in good to high yields. Subsequent iodocyclization afforded the corresponding 4-CF3-3-iodo-2-substituted thiophenes in good to high yields. It is noteworthy that the substrate bearing a triisopropylsilyl group was intact under optimal reaction conditions due to the bulky silyl group hindering the approach of electrophilic iodine [47] (Scheme 10).
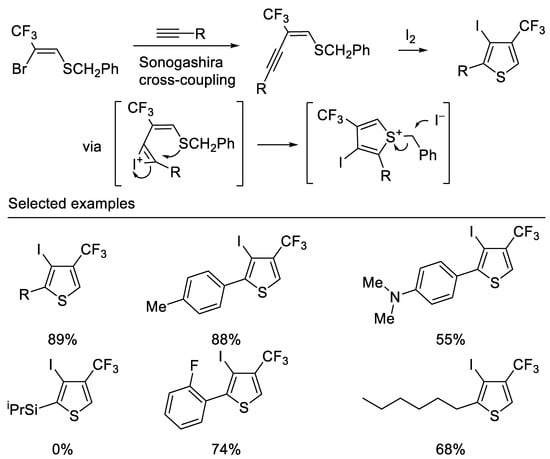
Scheme 10. Synthesis of trifluoromethylated thiophene derivatives.
Other methods to construct thiophene derivatives from β-CF3-1,3-enynes were also reported. In 2020, Song group developed a divergent strategy for the construction of 3-SCF2H-4-CF3-thiophenes from readily available 1,3-enynes and S8 via a tandem thiophene construction/selective C3 thiolation/difluoromethylthiolation under a ClCF2H atmosphere with excellent substrate compatibility. Experiments had shown that the construction of the thiophene ring may be a radical annulation process with S3•− generated in situ, and freon is used as a cheap difluoromethylation reagent. A series of 3-SeCF2H-4-CF3-selenophenes can also be constructed by similar strategies [48] (Scheme 11).
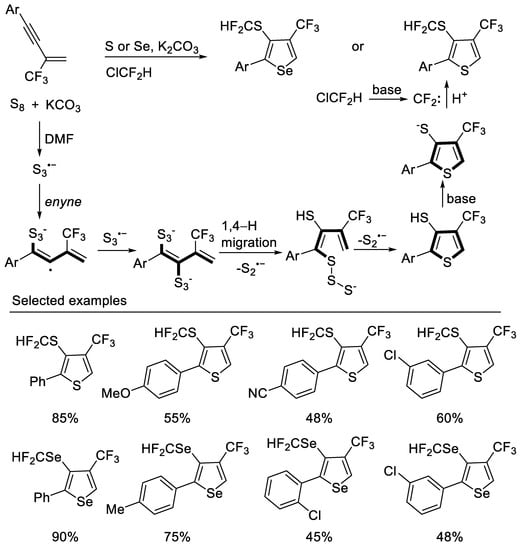
Scheme 11. Synthesis of 3-SCF2H-4-CF3-thiophenes derivatives.
In their subsequent work, they developed a divergent method for precise constructions of cyclic unsymmetrical diaryl disulfides or diselenides and polythiophenes from β-CF3-1,3-enynes and S8 when the ortho group is F, Cl, Br, and NO2 on aromatic rings. However, when the ortho group is H, disulfides (diselenides) were constructed These transformations undergo a cascade thiophene construction/selective C3-position thiolation process. A novel plausible radical annulation process was proposed and validated by DFT calculations [49] (Scheme 12).
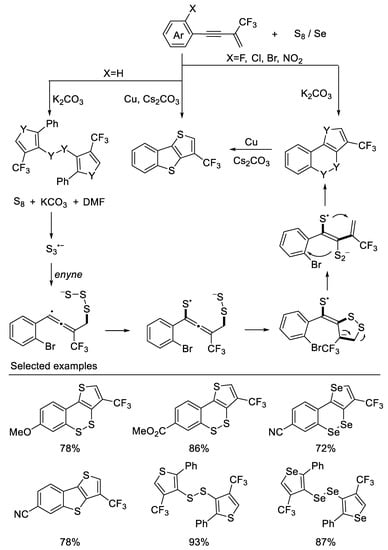
Scheme 12. Synthesis of diaryl disulfides or diselenides and polythiophenes derivatives.
In 2014, researchers developed novel divergent cyclizations of N-(2-(trifluoromethyl)-3-alkynyl)hydroxylamines which are easily prepared from the corresponding β-CF3-1,3- enynes and the salt of hydroxylamine under basic reaction conditions via simple nucleophilic addition by subtle choice of the catalyst system, leading to two important trifluoromethylated nitrogen containing heterocycles, such as 4-trifluoromethyl cyclic nitrone and pyrrole. The IPrAuNTf2/HNTf2 co-catalyzed cyclization of N-(2-perfluoroalkyl-3-alkynyl) hydroxylamines produces pyrroles in moderate to excellent yields, whereas the AgOTf-catalyzed reaction affords cyclic nitrones in high yields. The notable features of the method are its easily accessible starting materials, mild reaction conditions and divergent synthesis [50] (Scheme 13).
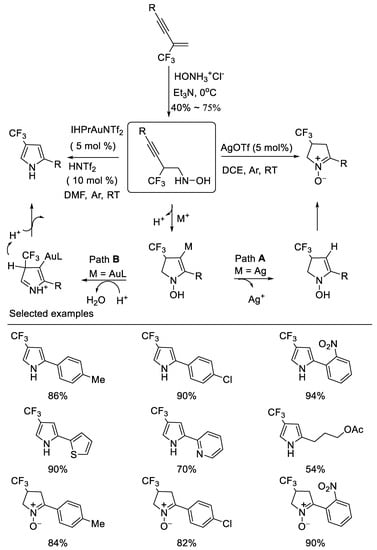
Scheme 13. Divergent synthesis of trifluoromethylated cyclic nitrone and pyrrole.
Following the above work, researchers subsequently developed a novel NIS-mediated oxidative cyclization of N-(2-trifluoromethyl-3-alkynyl) hydroxylamine under mild conditions, which provides a facile route to various 4-trifluoromethyl-5-acylisoxazoles. It was found that the NIS acts as both an oxidant and an electrophile for this sequential transformation. The key intermediate oxime formed by oxidation of hydroxylamine by NIS could be isolable, which underwent I+-induced O-selected 5-exo-dig cyclization and subsequent cascade reaction to afford 4-trifluoromethyl-5-acylisoxazoles. It is also noteworthy that no desired product was obtained when alkyne bearing TMS group or terminal alkyne was used as substrate [51] (Scheme 14a). Control experiments indicated that the oxygen atom of the ketone originated from water rather than from molecular dioxygen (Scheme 14b).
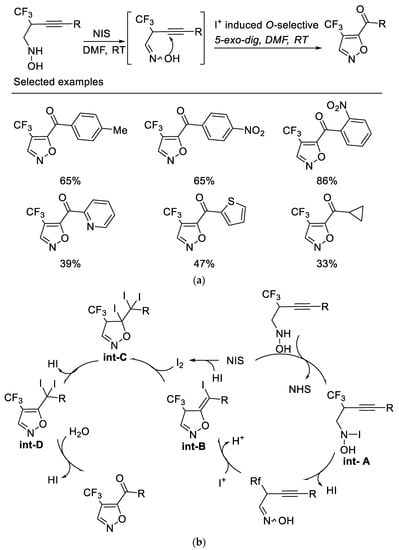
Scheme 14. (a) Synthesis of 4-trifluoromethyl-5-acylisoxazoles. (b) Plausible mechanism for synthesis of 4-trifluoromethyl-5-acylisoxazoles.
The exclusive O-selected 5-exo-dig cyclization of oxime in the above transformation conditions have aroused the interest because oximes can be employed as N-selective nucleophiles or as O-selective nucleophiles in many chemical transformations. researchers deduced that the Brønsted acid (HI) in situ formed in the above transformation conditions decreases the nucleophilicity of oxime nitrogen and destroys the inter- or intramolecular hydrogen bond between oxime oxygen and trifluoromethyl group, thus facilitating O-selective 5-exo-dig cyclization. researchers envisioned that with a proper choice of transition metal catalysts, the isolated oximes might undergo N-selective 5-endo-dig electrophilic cyclization owing to their inter- or intramolecular hydrogen bond between oxime oxygen and the trifluoromethyl group, which decreased the nucleophilicity of the oxime oxygen [52]. Thus, another type of interesting nitrogen containing heterocycle i.e., fluorinated N-hydroxypyrroles, could be obtained (Scheme 15, path a). Furthermore, if excess NIS and molecular iodine (I2) formed in situ in the reaction were reduced with a proper reductant, the in situ formed Brønsted acid would catalyze the O-selective 5-exo-dig or 6-endo-dig electrophilic cyclization of oximes; thus, an easy two-step, one-pot-synthesis of 4-trifluoromethyl-5-alkylisoxazoles (Scheme 15, path b) would be developed. Based on the above considerations, researchers developed the divergent regioselective cyclizations of N-(2-trifluoromethyl-3-alkynyl) oximes by subtle choice of gold(I) or Brønsted acid catalyst system, leading to 4-trifluoromethyl N-hydroxypyrroles or 5-akylisoxazoles. In order to avoid the tedious separation of unstable N-(2-trifluoromethyl-3-alkynyl) oximes, an easy two-step, one-pot synthesis of 4-trifluoromethyl-5-alkylisoxazoles from N-(2-trifluoromethyl-3-alkynyl) hydroxyl-amines is realized. This two-step, one-pot procedure is a complementary method for the synthesis of 4-trifluoromethyl-5-alkyl isoxazoles from those unstable N-(2-trifluoromethyl-3-alkynyl) oximes [53] (Scheme 15).
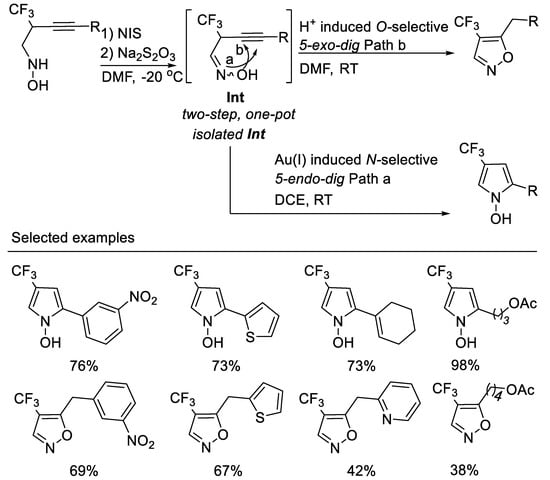
Scheme 15. Divergent synthesis of trifluoromethylated N-hydroxypyrroles or 5-akylisoxazoles.
After researchers studied the chemistry of β-CF3-1,3-enynes with the bisnucleophile hydroxylamine and subsequent transformations, researchers then studied the reaction of β-CF3-1,3-enynes with the bisnucleophile 2-aminomalonates. A dramatic substituent effect was observed in the reaction of β-CF3-1,3-enynes with the bisnucleophile 2-aminomalonates. When N-tosylated 2-aminomalonate was used as bisnucleophile, the reactions proceeded smoothly to afford 2-fluoro-2-pyrrolines via double direct C-F substitutions [53]. In contrast, either 4-trifluoromethyl pyrrolidines or gem-difluoro-1,3- conjugated enynes were delivered when N-acetylated 2-aminomalonate was used as reaction partner. β-CF3-1,3-enynes show an interesting substituent effect on the product diversity. β-CF3-1,3-enynes bearing electron-donating or weak electron-withdrawing groups, such as Me, MeO, Cl and Br, on the aryl substituent of the alkyne moiety afford functionalized gem-difluoro-1,3-conjugated enynes in moderate to good yields, whereas 4-trifluoromethyl pyrrolidines are isolated as the predominant product in moderate to good yields from those β-CF3-1,3-enynes with strong electron-deficient aromatic substituent. Various functionalized 4-(difluoromethylene)-1,2,3,4-tetrahydropyridines could be obtained in good yields via the gold(I)-catalyzed intramolecular 6-endo-dig cyclization of the corresponding gem-difluoro-1,3-conjugated enynes under mild conditions [54] (Scheme 16).
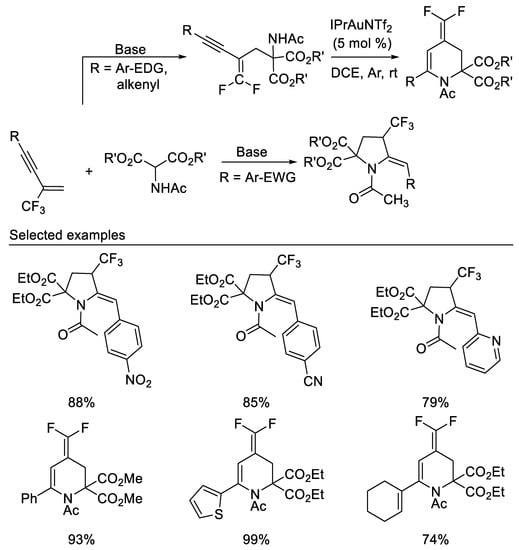
Scheme 16. Divergent synthesis of trifluoromethylated pyrrolidines or difluoromethylidene tetrahydropyridines.
In 2017, researchers developed the first example of tandem intermolecular hydroamination and cyclization reaction of β-CF3-1,3-enynes with bisnucleophile primary amines affording 4-trifluoromethyl-3-pyrrolines by employing a cheap silver catalyst under mild reaction conditions. This new method is compatible with alkyl, aryl, and allyl primary amines, representing an atom-economic protocol for the construction of 4-trifluoromethyl-3-pyrrolines for the first time. It should be noted that the reaction also works well for aromatic primary amines, albeit requiring a higher reaction temperature [55] (Scheme 17).
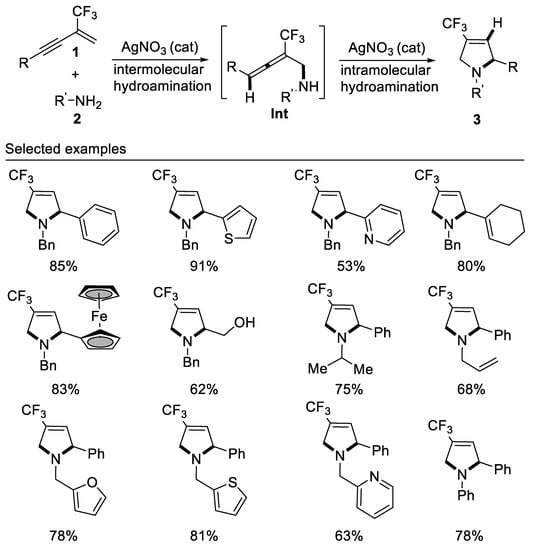
Scheme 17. Synthesis of 4-trifluoromethyl-3-pyrrolines.
Following the above work, researchers subsequently developed a facile two-step, one-pot method for the synthesis of a range of halogenated trifluoromethylated pyrroles from β-CF3-1,3-enynes, readily aliphatic primary amines and halogenating agents, such as NBS and NIS. By variation of the halogenating agents, ring trifluoromethylated monoiodo pyrrole or dibromo pyrrole skeletons can be readily accessed in moderate to good yields. The different outcome of the reactions with NIS and NBS may be due to their different electrophilic properties towards pyrrole. This two-step, one-pot method employs a key halogenating-agents mediating step to trigger a cascade process featuring an initial electrophilic cyclization of the first intermolecular hydroamination product [56] (Scheme 18).
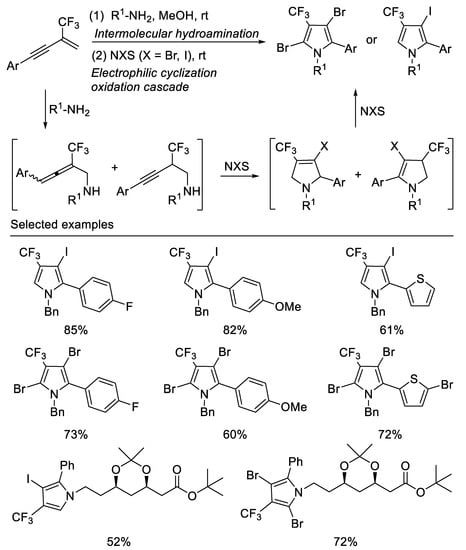
Scheme 18. Synthesis of halogenated trifluoromethylated pyrroles.
More recently, in 2021, researchers developed the first example of a tandem double hydroamination reaction of β-CF3-1,3-enynes with bisnucleophiles hydrazine derivatives under mild reaction conditions. By variation of the substituents on the hydrazine nitrogen atom, three types of trifluoromethylated pyrazolidines, pyrazolines and pyrazoles can be readily accessed in moderate to good yields. The reaction with simple hydrazine monohydrate or sulfonyl hydrazines as nucleophiles produces 1,3,4-trisubstituted pyrazolines, whereas the reaction with acetyl hydrazine as nucleophiles affords 1,4,5-trisubstituted pyrazolidines. Using phenylhydrazine or tert-butylhydrazine as reaction partners, the products are easily oxidized to form 1,4,5-trisubstituted pyrazoles [57] (Scheme 19).
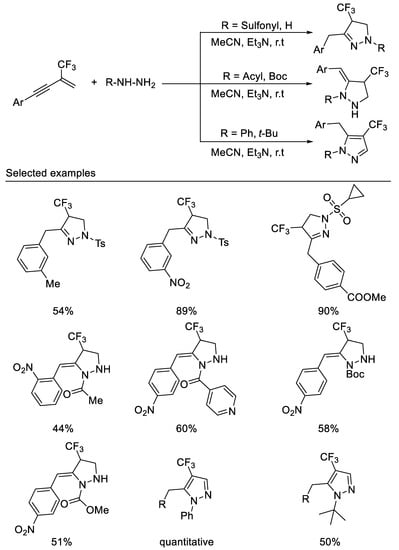
Scheme 19. Divergent synthesis of trifluoromethylated pyrazolidines, pyrazolines and pyrazoles.
3.2. Construction of Six-Membered Trifluoromethylated Heterocycles
In 2000, the Qing group first reported the synthesis of 4-trifluoromethyl-2H-pyrans by palladium-catalyzed cyclization of (E)-3-alkynyl-3-trifluoromethyl allylic alcohols. The 6-endo-dig cyclization, not the 5-exo-dig cyclization, is favored due to the fact that the trifluoromethyl group possesses powerful electron-withdrawing properties and (E)-3-alkynyl-3-trifluoromethyl allylic alcohols bearing an aryl group adjacent to the hydroxyl lead to dienone under same reaction conditions [58] (Scheme 20).
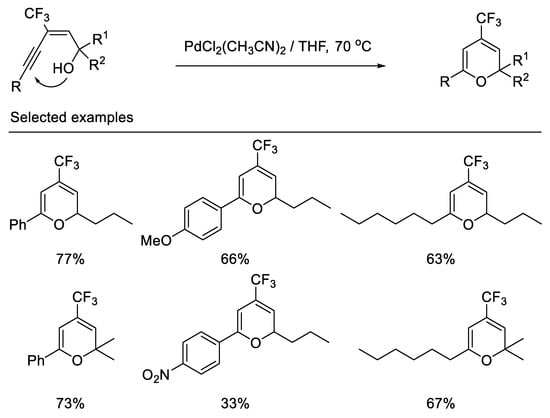
Scheme 20. Synthesis of 4-trifluoromethyl-2H-pyrans derivatives.
A straightforward and efficient approach to alkyne-functionalized ring-monofluorinated 4H-pyrans via a simple base-mediated cascade reaction of β-CF3-1,3-enynes with 1,3-dicarbonyl compounds or monocyano- substituted carbon nucleophiles, such as 3-oxo-3-phenylpropanenitrile, 3-oxo-butyronitrile was developed by a group [59] and Chang [10]. Substituted alkynyl group was used as an activating group. The key events of this reaction involve two consecutive C-F substitutions under very mild conditions (Scheme 21).
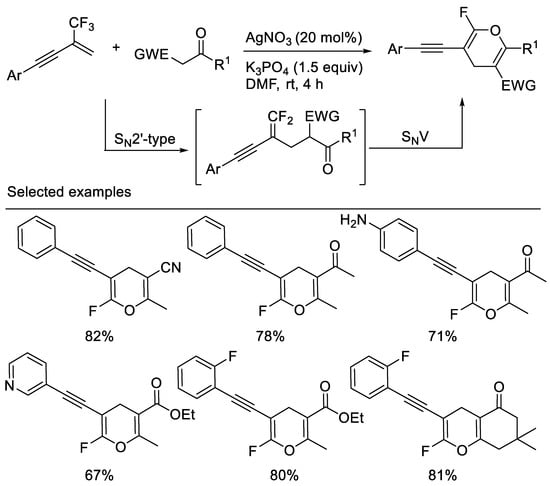
Scheme 21. Synthesis of alkyne-functionalized ring-monofluorinated 4H-pyrans.
Very recently, researchers developed the first example of a tandem intermolecular hydrocarbonation/intramolecular heterocyclization reaction of β-CF3-1,3-enynes with bisnucleophiles β -ketothioamides under mild reaction conditions. By variation of the substituents linked to the carbonyl or on the β-ketothioamides nitrogen atom, ring trifluoromethylated pyrans, or thiopyrans, can be readily accessed in moderate to good yields. Enynes possessing electron-withdrawing aryl groups on the alkyne moiety are generally good candidates for present transformation and β-ketothioamides bearing a piperidine substituent on the amide moiety, and (hetero)aryl groups on the keto moiety would mainly afford pyran, whereas β-ketothioamides bearing pyrrolidine substituent on the amide moiety and (hetero)aryl or alkyl groups on keto moiety lead to the formation of thiopyrans. Other substituted forms of N,N-disubstituted β-ketothioamides would give mixtures of pyrans and thiopyrans. researchers think that the formation of the oxygen enol or sulfur enol intermediate Int-B could be affected by electronic and spatial effects of substituents on either the keto moiety or the nitrogen atom of the β-ketothioamide from the reaction of Int-A under basic conditions, which results in pyrans or thiopyrans. β-ketothioamides bearing a piperidine substituent on the amide moiety and (hetero)aryl groups on the keto moiety would mainly form oxyenols, leading to the formation of pyran, whereas sulfur enol would predominantly form β-ketothioamides bearing a pyrrolidine substituent on the amide moiety and (hetero)aryl or alkyl groups on the keto moiety, leading to the formation of thiopyrans. Other substituted forms of N,N-disubstituted β-ketothioamides would give mixtures of oxygen enol and sulfur enol which result in mixtures of pyrans and thiopyrans. The salient features of this tandem include atom-economical, mild reaction condition, ease of operation and product diversity [60] (Scheme 22).
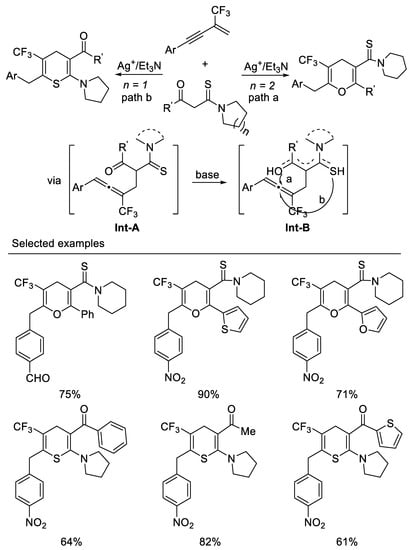
Scheme 22. Synthesis of ring trifluoromethylated pyrans, or thiopyrans.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27249020
References
- Meanwell, N.A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591.
- Wang, J.; Sánchez-Rosellό, M.; Aceňa, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506.
- Bassetto, M.; Ferla, S.; Pertusati, F. Polyfluorinated groups in medicinal chemistry. Future Med. Chem. 2015, 7, 527–546.
- Gouverneur, V.; Müller, K. Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications; Imperial College Press: London, UK, 2012.
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013.
- Boltalina, O.V.; Nakajima, T. New Fluorinated Carbons: Fundamentals and Applications; Elsevier: Amsterdam, The Netherlands, 2016.
- Lowe, P.T.; O’Hagan, D. 4’-Fluoro-nucleosides and nucleotides: From nucleocidin to an emerging class of therapeutics. Chem. Soc. Rev. 2023, in press.
- Zhang, C.; Yan, K.; Fu, C.; Peng, H.; Hawker, C.J.; Whittaker, A.K. Biological Utility of Fluorinated Compounds: From Materials Design to Molecular Imaging, Therapeutics and Environmental Remediation. Chem. Rev. 2022, 122, 167–208.
- He, J.; Li, Z.; Dhawan, G.; Zhang, W.; Sorochinsky, A.E.; Butler, G.; Soloshonok, V.A.; Han, J. Fluorine-containing drugs approved by the FDA in 2021. Chin. Chem. Lett. 2023, 34, 107578–107587.
- Han, J.; Kiss, L.; Mei, H.; Remete, A.M.; Ponikvar-Svet, M.; Sedgwick, D.M.; Roman, R.; Fustero, S.; Moriwaki, H.; Soloshonok, V.A. Chemical Aspects of Human and Environmental Overload with Fluorine. Chem. Rev. 2021, 121, 4678–4742.
- Firouzjaei, M.D.; Zolghadr, E.; Ahmadalipour, S.; Taghvaei, N.; Afkhami, F.A.; Nejati, S.; Elliott, M.A. Chemistry, abundance, detection and treatment of per-and polyfuoroalkyl substances in water: A review. Environ. Chem. Lett. 2022, 20, 661–679.
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current Contributions of Organofluorine Compounds to the Agrochemical Industry. iScience 2020, 23, 101467–101489.
- Furuya, T.; Kamlet, A.S.; Ritter, T. Catalysis for fluorination and trifluoromethylation. Nature 2011, 473, 470–477.
- Tomashenko, O.A.; Grushin, V.V. Aromatic trifluoromethylation with metal complexes. Chem. Rev. 2011, 111, 4475–4521.
- Zhang, C.-P.; Chen, Q.-Y.; Guo, Y.; Xiao, J.-C.; Gu, Y.-C. Progress in fluoroalkylation of organic compounds via sulfinatodehalogenation initiation system. Chem. Soc. Rev. 2012, 41, 4536–4559.
- Yang, X.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015, 115, 826.
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of fluorine and fluorinecontaining functional groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264.
- Tlili, A.; Toulgoat, F.; Billard, T. Synthetic Approaches to Trifluoromethoxy-Substituted Compounds. Angew. Chem. Int. Ed. 2016, 55, 11726–11735.
- Rong, J.; Ni, C.; Hu, J. Metal-Catalyzed Direct Difluoromethylation Reactions. Asian J. Org. Chem. 2017, 6, 139–152.
- Liu, Q.; Ni, C.; Hu, J. China’s flourishing synthetic organofluorine chemistry: Innovations in the new millennium. Natl. Sci. Rev. 2017, 4, 303–325.
- Yang, L.; Dong, T.; Revankar, H.M.; Zhang, C.-P. Recent progress on fluorination in aqueous media. Green Chem. 2017, 19, 3951–3992.
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon-Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964.
- Szpera, R.; Moseley, D.F.J.; Smith, L.B.; Sterling, A.J.; Gouverneur, V. The Fluorination of C-H Bonds: Developments and Perspectives. Angew. Chem. Int. Ed. 2019, 58, 14824–14848.
- Sicard, A.J.; Baker, R.T. Fluorocarbon Refrigerants and their Syntheses: Past to Present. Chem. Rev. 2020, 120, 9164–9303.
- Linclau, B.; Ardá, A.; Reichardt, N.C.; Sollogoub, M.; Unione, L.; Vincent, S.P.; Jiménez-Barbero, J. Fluorinated carbohydrates as chemical probes for molecular recognition studies. Current status and perspectives. Chem. Soc. Rev. 2020, 49, 3863–3888.
- Tarantino, G.; Hammond, C. Catalytic C(sp3)-F bond formation: Recent achievements and pertaining challenges. Green Chem. 2020, 22, 5195–5209.
- Huonnic, K.; Linclau, B. The Synthesis and Glycoside Formation of Polyfluorinated Carbohydrates. Chem. Rev. 2022, 122, 15503–15602.
- Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 2011, 111, 455–529.
- Dherbassy, Q.; Manna, S.; Talbot, F.J.T.; Prasitwatcharakorn, W.; Perry, G.J.P.; David, J. Procter, D.J. Copper-catalyzed functionalization of enynes. Chem. Sci. 2020, 11, 11380–11393.
- Mlostoń, G.; Shermolovich, Y.; Heimgartner, H. Synthesis of Fluorinated and Fluoroalkylated Heterocycles Containing at Least One Sulfur Atom via Cycloaddition Reactions. Materials 2022, 15, 7244.
- Metelev, V.G.; Bogdanov, A.A., Jr. Synthesis and applications of theranostic oligonucleotides carrying multiple fluorine atoms. Theranostics 2020, 10, 1391–1414.
- Ogiwara, Y.; Sakai, N. Acyl Fluorides in Late-Transition-Metal Catalysis. Angew. Chem. Int. Ed. 2020, 59, 574–594.
- Wehbi, M.; Mehdi, A.; Negrell, C.; David, G.; Alaaeddine, A.; Améduri, B. Phosphorus-Containing Fluoropolymers: State of the Art and Applications. ACS Appl. Mater. Interfaces 2020, 12, 38–59.
- Butcher, T.W.; Amberg, W.M.; Hartwig, J.F. Transition-Metal-Catalyzed Monofluoroalkylation: Strategies for the Synthesis of Alkyl Fluorides by C-C Bond Formation. Angew. Chem. Int. Ed. 2022, in press.
- Ai, H.J.; Ma, X.; Song, Q.; Wu, X.F. C–F bond activation under transition-metal-free conditions. Sci. China Chem. 2021, 64, 1630–1659.
- Zhang, B.; Wang, J. Assembly of versatile fluorine-containing structures via N-heterocyclic carbene organocatalysis. Sci. China Chem. 2022, 65, 1691–1703.
- Chen, S.; Zhang, J.; Yang, M.; Liu, F.; Xie, Z.; Liu, Y.; Lin, W.; Wang, D.; Li, X.; Wang, J. Diastereoselective synthesis of cyclopropanes bearing trifluoromethyl-substituted all-carbon quaternary centers from 2-trifluoromethyl-1,3-enynes beyond fluorine elimination. Chem. Commun. 2019, 55, 3879–3882.
- Chen, G.-S.; Yan, X.-X.; Chen, S.-J.; Mao, X.-Y.; Li, Z.-D.; Liu, Y.-L. Diastereoselective Synthesis of 1,3-Diyne-Tethered Trifluoromethylcyclopropanes through a Sulfur Ylide Mediated Cyclopropanation/DBU-Mediated Epimerization Sequence. J. Org. Chem. 2020, 85, 6252–6260.
- Ma, Z.; Deng, Y.; He, J.; Cao, S. Solvent-controlled base-free synthesis of bis(trifluoromethyl)-cyclopropanes and –pyrazolines via cycloaddition of 2-trifluoromethyl-1,3-enynes with 2,2,2-trifluorodiazoethane. Org. Biomol. Chem. 2022, 20, 5071–5075.
- Hwang, J.H.; Jung, Y.H.; Hong, Y.Y.; Jeon, S.L.; Jeong, I.H. Synthesis of novel 2-trifluoromethyl-1-methylene-3-phenylindene derivatives via carbocyclization reaction of 2-trifluoromethyl-1,1-diphenyl-1,3-enynes. J. Fluor. Chem. 2011, 132, 1227–1231.
- Trost, B.M.; Laurent Debien, L. Palladium-Catalyzed Trimethylenemethane Cycloaddition of Olefins Activated by the σ-Electron-Withdrawing Trifluoromethyl Group. J. Am. Chem. Soc. 2015, 137, 11606–11609.
- Zeng, Y.; Huang, C.; Ni, P.; Liu, L.; Xiao, Y.; Zhang, J. Silver-Catalyzed Double Hydrocarbonation of 2-Trifluoromethyl-1,3-Conjugated Enynes with 1,3-Dicarbonyl Compounds: Synthesis of Ring-Trifluoromethylated Cyclopentene. Adv. Synth. Catal. 2017, 359, 3555–3559.
- Zhang, J.; Ma, Z.-G.; Tian, Y.; Li, W.; Gao, W.-C.; Chang, H.-H. Divergent Synthesis of Fluorinated Alkenes, Allenes, and Enynes via Reaction of 2-Trifluoromethyl-1,3-enynes with Carbon Nucleophiles. J. Org. Chem. 2022, 87, 15086–15100.
- Chen, S.-J.; Chen, G.S.; Zhang, J.-W.; Li, Z.-D.; Zhao, Y.-L.; Liu, Y.-L. Phosphine-catalyzed cycloadditions of trifluoromethyl enynes/enediynes with allenoates: Access to cyclopentenes containing a CF3-substituted quaternary carbon center. Org. Chem. Front. 2020, 7, 3399–3405.
- Zatolochnaya, O.V.; Gevorgyan, V. Synthesis of Fluoro- and Perfluoroalkyl Arenes via Palladium-Catalyzed Benzannulation Reaction. Org. Lett. 2013, 15, 2562–2565.
- Yang, Z.-J.; Hu, B.-L.; Deng, C.-L.; Zhang, X.-G. Iron-Promoted Electrophilic Annulation of Aryl Enynes with Disulfides or Diselenides Leading to Polysubstituted Naphthalenes. Adv. Synth. Catal. 2014, 356, 1962–1966.
- Ikeda, G.; Kawasaki, M.; Hanamoto, T. Sonogashira cross-coupling of (Z)-2-bromo-2-CF3-vinyl benzyl sulfide and access to 4-CF3-3-iodo-2-substituted thiophenes. J. Fluor. Chem. 2021, 248, 109838–109843.
- Jin, S.; Kuang, Z.; Song, Q. Precise Construction of SCF2H or SeCF2H Groups on HeteroarenesGenerated in Situ from CF3-Containing 1,3-Enynes. Org. Lett. 2020, 22, 615–619.
- Jin, S.; Li, S.-J.; Ma, X.; Su, J.; Chen, H.; Lan, Y.; Song, Q. Elemental-Sulfur-Enabled Divergent Synthesis of Disulfides, Diselenides, and Polythiophenes from β-CF3-1,3-Enynes. Angew. Chem. Int. Ed. 2021, 60, 881–888.
- Zeng, Q.; Zhang, L.; Yang, J.; Xu, B.; Xiao, Y.; Zhang, J. Pyrroles versus cyclic nitrones: Catalyst-controlled divergent cyclization of N-(2-perfluoroalkyl-3-alkynyl) hydroxylamines. Chem. Commun. 2014, 50, 4203–4206.
- Zhang, L.; Zeng, Q.; Mao, A.; Wu, Z.; Luo, T.; Xiao, Y.; Zhang, J. NIS-mediated oxidative cyclization of N-(2-trifluoromethyl-3-alkynyl) hydroxylamines: A facile access to 4-trifluoromethyl-5-acylisoxazoles. Org. Biomol. Chem. 2014, 12, 8942–8946.
- Tan, B.; Shi, Z.; Chua, P.; Li, Y.; Zhong, G. Unusual domino Michael/Aldol condensation reactions employing oximes as N-selective nucleophiles: Synthesis of N-hydroxypyrroles. Angew. Chem. Int. Ed. 2009, 48, 758–761.
- Yang, J.; Zhou, X.; Zeng, Y.; Huang, C.; Xiao, Y.; Zhang, J. Synthesis of 2-fluoro-2-pyrrolines via tandem reaction of α-trifluoromethyl-α,β-unsaturated carbonyl compounds with N-tosylated 2-aminomalonates. Chem. Commun. 2016, 52, 4922–4925.
- Yang, J.; Zhou, X.; Zeng, Y.; Huang, C.; Xiao, Y.; Zhang, J. Divergent synthesis from reactions of 2-trifluoromethyl-1,3-conjugated enynes with N-acetylated 2-aminomalonates. Org. Biomol. Chem. 2017, 15, 2253–2258.
- Zhou, X.; Huang, C.; Zeng, Y.; Xiong, J.; Xiao, Y.; Zhang, J. Silver-catalysed tandem hydroamination and cyclization of 2-trifluoromethyl-1,3-enynes with primary amines: Modular entry to 4-trifluoromethyl-3-pyrrolines. Chem. Commun. 2017, 53, 1084–1087.
- Huang, C.-Q.; Zeng, Y.; Cheng, H.; Hu, A.; Liu, L.; Xiao, Y.; Zhang, J. A One-Pot Construction of Halogenated Trifluoromethylated Pyrroles through NXS (X = Br, I) Triggered Cascade. Org. Lett. 2017, 19, 4968–4971.
- Wei, L.; Ding, S.; Liu, M.; Yu, Z.; Xiao, Y. Synthesis of Trifluoromethylated Pyrazolidines, Pyrazolines and Pyrazoles via Divergent Reaction of β-CF3-1,3-Enynes with Hydrazines. Org. Lett. 2021, 23, 7718–7723.
- Qing, F.-L.; Gao, W.-Z. The first synthesis of 4-trifluoromethyl-2H-pyrans by palladium-catalyzed cyclization of (E)-3-alkynyl-3-trifluoromethyl allylic alcohols. Tetrahedron Lett. 2000, 41, 7727–7730.
- Yang, J.; Mao, A.; Yue, Z.; Zhu, W.; Luo, X.; Zhu, C.; Xiao, Y.; Zhang, J. A simple base-mediated synthesis of diverse functionalized ring-fluorinated 4H-pyrans via double direct C−F substitutions. Chem. Commun. 2015, 51, 8326.
- Zeng, Q.; Huang, X.; Liu, M.; Yu, Z.; Xiao, Y. Synthesis of Trifluoromethylated 4H-Pyran and 4H-Thiopyran via Divergent Reaction of β-CF3-1,3-Enynes with Ketothioamides. Org. Lett. 2022, 24, 8186–8191.
This entry is offline, you can click here to edit this entry!