Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Na/K-ATPase (NKA), a large transmembrane protein, is expressed in the plasma membrane of most eukaryotic cells. It maintains resting membrane potential, cell volume and secondary transcellular transport of other ions and neurotransmitters.++
- Na+/K+-ATPase
- neurodegenerative diseases
- protein–protein interaction
1. 简介
神经退行性疾病(NDDs)是一组以特定脑区慢性、进行性和不可逆神经元丢失为特征的疾病[1]。大脑中神经元的耗竭伴随着受影响个体的身体、心理社会、情绪和认知功能障碍[2]。NDD包括阿尔茨海默病(AD)、帕金森病(PD)、肌萎缩侧索硬化症(ALS)、亨廷顿病(HD)、多发性硬化症(MS)等[3]。随着平均年龄和预期寿命的增加,NDD的发病率也会增加[4]。然而,没有改善疾病的疗法来减缓NDD的进展。
在实验性NDD中发现NKA功能障碍[5,[5]。恢复NKA的活性和膜稳定性可以缓解神经系统残疾[6]。此外,NKA功能障碍还与NDD的各种病理特性有关,例如错误折叠和聚集的蛋白质,线粒体功能障碍和氧化应激[7]。此外,我们最近的研究表明,稳定NKA的单克隆抗体DR5-12D可以稳定膜NKA并保护大脑免受α-syn诱导的病理[8]。简而言之,NKA可能作为NDD的一种有前途的治疗方式。
2. 钠/钾-ATP酶++
2.1. NKA 的发现
动作电位是可兴奋细胞膜中的一种电现象,其传播信号而不会衰减[9]。艾伦·劳埃德·霍奇金爵士和安德鲁·菲尔丁·赫胥黎爵士发现了它的机制,然后分享了1963年的诺贝尔奖[10]。科学家假设哺乳动物细胞需要“钠泵”来产生动作电位的离子梯度[11]。在发现动作电位一个多世纪后,丹麦生物化学家Jens Christian Skou发现了一种新的ATP酶,该酶依赖于Na和K的存在。此外,这种ATP酶可能被ouabain(钠泵抑制剂)特异性抑制,证明它是“钠泵”[12]。因此,Skou和另外两位科学家因其在ATP酶方面的工作而分享了1997年诺贝尔化学奖[13]。++
2.2. NKA的结构、亚单位和分布
NKA属于P型ATP酶家族,具有酰基磷酸中间体。NKA包含3个不同的亚基,α,β和γ(FXYD),具有等摩尔化学计量[14]。图 1 说明了 NKA 的结构。α亚基在N末端有10个跨膜区域和3个胞质结构域:N结构域(核苷酸结合)、P结构域(磷酸化)和A结构域(执行器)[15]。由于它含有ATP结合位点,因此α亚基也是催化亚基[16,[16]。α亚基有四种亚型:α1-α4。α1在所有哺乳动物组织中普遍表达,其突变是致命的;α2主要在脑神经胶质细胞中表达,其突变引起多种疾病,如家族性偏瘫性偏头痛2型(FHM2)、交替性儿童偏瘫1型(AHC1)和癫痫[18,[17];α3在脑中具有神经元特异性,其突变可引起快速发作的肌张力障碍-帕金森综合征(RDP)、小脑共济失调、感音神经性听力损失(CAPOS)等[16,20,21,22,[18];α4仅在睾丸中表达,其稳定性对雄性繁殖力至关重要[19]。
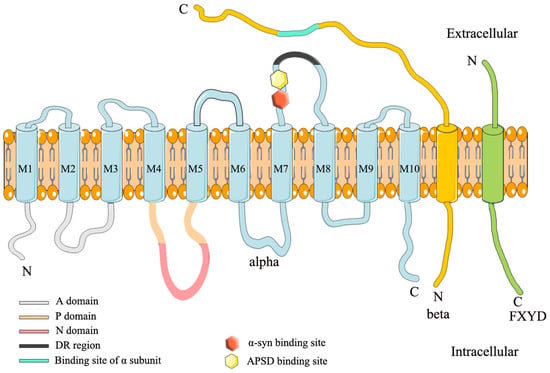
β亚基是单个跨膜糖蛋白,小于α亚基[21]。虽然它不形成NKA的转运孔,但β亚基会影响NKA动力学。作为分子伴侣,它有助于NKA的正确组装和膜递送,并保持细胞粘附和细胞极性[28,29,30,[22]。β亚基有四种亚型:β1-β4。β1广泛表达,而β2在大脑中的神经胶质细胞,微血管和脉络丛中表达。β3在大脑中的微粒体中表达[21]。
γ亚基(也称为FXYD蛋白)不是酶活性所必需的,但它调节NKA的催化特性[32,[23]。在哺乳动物中,FXYD亚基包括7种亚型,其中FXYD2是调节NKA的第一个亚基[24]。α、β和γ亚基的多种亚型组合显示出独特的NKA活性,可以满足不同组织和细胞的需求[11]。
2.3. NKA 的功能
作为一种泵蛋白,NKA调节Na和K的离子梯度以确定静息膜电位[25]。此外,离子梯度会影响渗透压、细胞体积,并为神经递质和其他离子的二次转运提供能量[36,[26]。此外,NKA还与其他蛋白质形成大分子复合物,以触发许多信号通路[38,[27]。例如,在AD啮齿动物模型中,NKA与不同的淀粉样蛋白β肽结合,诱导神经元丢失和血管功能障碍[28]。++
3. NKA在阿尔茨海默病中的作用
阿尔茨海默病(AD)是一种不可逆的严重神经退行性疾病,由Alois Alzheimer于1907年首次描述[29]。AD患者存在认知缺陷、记忆功能障碍和情绪障碍[30]。AD的特征是脑中积聚细胞外淀粉样蛋白β斑(Aβ)斑块和细胞内tau神经原纤维缠结[31]。AD的危险因素包括年龄、家族史、性别、教育程度和合并症[44,45,[32]。累积证据表明,AD患者脑部和AD啮齿动物模型中NKA活性降低,提示NKA可能与AD的发生有关[47,[33]。
3.1. NKA 和 Aβ 斑块
Aβ斑块的存在是AD的重要特征,Aβ斑块主要由Aβ肽组成[34]。它们表现为肽的产生、清除和聚集之间的不等效平衡,这导致Aβ肽积累[35]。Aβ前体蛋白(APP)被β分泌酶和γ分泌酶裂解产生Aβ肽[36]。APP也可能被α分泌酶和γ分泌酶切割,产生可溶性淀粉样蛋白前体蛋白α(sAPPα)[37]。有趣的是,NKA作为Aβ肽和sAPPα的调节剂来调节AD中的学习和记忆[37]。最近的研究表明,sAPPα在神经元表面招募了NKAα3的簇。sAPPα和NKAα3的相互作用调节了胞内Na和Ca的水平+2+ that is required for APP to reach the cell surfaces. When APP interacted with sAPPα in the membrane, the APP/sAPPα complex triggered a cascade of events promoting sAPPα-induced axonal outgrowth [37]. However, the expression levels of sAPPα and NKA were significantly decreased in an AD rodent model compared with the control group [38]. The sAPPα deficiency reduced the neuroprotective effect of sAPPα. In contrast, the activity and expression level of β-secretase were increased in AD patients and rodent models, which produced more Aβ peptides [39]. The overproduction of Aβ peptides influences the activity and expression level of NKA through downstream pathways.
3.1.1. Interactions between NKA and Aβ Peptides
Aβ peptides form structurally distinct complexes to exert different toxic functions via different targets [40]. Specifically, amylospheroids (ASPDs) are a population of neurotoxic Aβ aggregates containing approximately 30 Aβ monomers [41]. In a normal physiological condition, full-length APP is either cleaved by β- and γ-secretase cleavages to produce Aβ aggregates or re-internalized via an endosomal/lysosomal degradation pathway [42]. Evidence showed that pharmacological inhibition of proteasome-associated degradation of APP dramatically upregulated intra-neuronal ASPD levels [42] (Figure 2). Then, the ASPD-producing neurons died non-apoptotically. More importantly, ASPDs were secreted and caused the degeneration of adjacent NKAα3-expressing neurons [42]. Compelling studies indicated that NKAα3 was the neuronal death-inducing target of ASPD. The region in the fourth extracellular loop (Ex4) of NKAα3 encompasses residues Asn879 and Trp880. These residues were found to be essential for ASPD/NKAα3 interaction [23]. It is worth mentioning that the ASPD/NKAα3 complex impaired NKAα3-specific activity; activated N-type voltage-gated calcium channels; and caused mitochondrial calcium dyshomeostasis, tau abnormalities and neurodegeneration [23]. In addition to neuropathological features, 60–90% of AD patients exhibit vascular dysfunction, which may precede the onset of AD [43]. In in vitro blood cell cultures and ex vivo blood vessels, ASPDs bound to NKAα3 in endothelial cells, activating protein kinase C (PKC). The ASPD/NKAα3 complex increased the PKC-phosphorylated inactive form of endothelial nitric oxide synthase (eNOS) and decreased nitric oxide (NO) production. NO deficiency suppressed the relaxation of blood microvessels and might cause a reduction in cerebral blood flow and other vascular dysfunctions in mouse models of AD [43]. As stated above, these observations revealed the mechanisms of Aβ-induced NKAα3 impairment in AD.
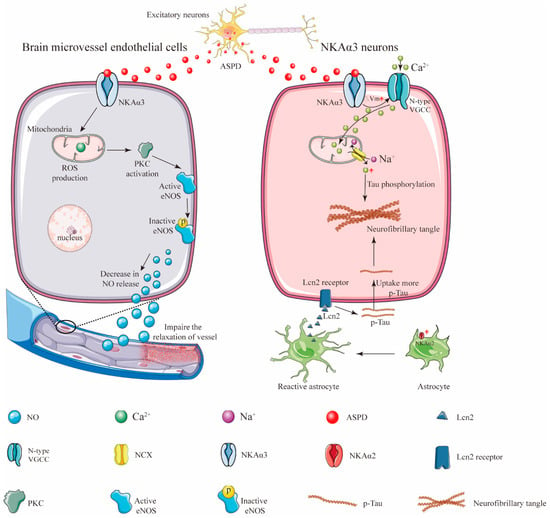
Figure 2. Schematic illustration showing how ASPD secretion from the excitatory neurons induces neuronal loss and vascular pathologies. In response to central nervous system (CNS) damage, astrocytic NKAα2 level was upregulated and contributed to astrogliosis [44]. The activated astrocytes then secreted more proinflammatory protein lipocalin-2 (Lcn-2) to neurons, causing neurons to uptake more extracellular tau and resulting in the formation of the neurofibrillary tangles [44].
However, ASPD do not exist at the early stages of AD and cannot damage NKA at these stages of the disease [28]. Intriguingly, some recent studies conducted at the early stages of AD demonstrated that NKAα1 also bound to monomeric Aβ42 to form a tight, equimolar complex. The Aβ42/NKAα1 complex disrupted NKA function and activated Src-kinase in neuroblastoma cells [28]. These findings support the notion that Aβ42 is a putative regulator of NKA. In line with this, some studies revealed that the phosphorylation of Aβ42 at Ser8 (pS8-Aβ) might neutralize some pathogenic properties of Aβ, reverse NKA activity by preventing the binding of Aβ42 to NKA, and then reduce cerebral plaque deposition [28]. These amazing and thrilling studies unveil a new outlook towards NKA structural derivatives to prevent the “protein–NKA” interaction for the treatment of AD.
3.1.2. NKA and Oxidative Stress
Aβ deposition caused oxidative stress in neuronal cells, which resulted in severe cell damage in AD patients and rodent models [45]. Methionine at residue 35 (Met35) of the Aβ sequence was required for Aβ-induced oxidative damage in AD [46]. Additional studies indicated that trans fatty acids enhance Aβ-induced oxidative stress in PC12 cells [46]. Following studies found that trans fatty acids reacted with Met35 of the Aβ residue. This complex significantly impaired the activity of NKA via enhancing the generation of ROS and nitric oxide and elevating caspase-3, caspase-8 and nitric oxide synthase activities [46]. Similarly, some studies found that the glutathionylation of the NKA α subunit determined enzyme the redox sensitivity. Meanwhile, glutathionylation of the NKA α subunit depended on the redox status of cells during the enzyme biosynthesis [47]. It is interesting to observe that long-term exposure to Aβ changed the thiol redox status of SH-SY5Y cells and inhibited NKA activity by upregulating the glutathionylation of the α subunit of NKA [41]. These studies indicate that NKA deficiency observed in AD may, at least in part, stem from Aβ-induced oxidative stress. Consistently, some antioxidants, such as 17β-estradiol, genistein, basic fibroblast growth factor (bFGF) and organosulfur compounds exerted their neuroprotective effects by restoring NKA activity in AD rodent models [48][49][50][51]. Thus, the drugs targeted at restoring NKA activity may be a potential therapeutic target in oxidative stress-induced AD.
3.2. NKA and Tau
As a microtubule-associated protein, tau affects the assembly and stabilization of the microtubular cytoskeleton [52]. The pathological form of tau is the principal component of paired helical filaments (PHFs) which are further associated with neurofibrillary tangles (NFTs). NFTs are the main hallmarks of AD [52]. Numerous studies reported that tau aggregates could be released from one and spread to other neurons, displaying “prion-like” properties [53]. Evidence showed that exogenous fibrillar tau (Fib-tau) formed clusters on the neuronal surfaces. Fib-tau clusters destabilized NKAα3 and formed a complex with NKAα3. This complex reduced neurons’ capacity to control membrane depolarization and exacerbated neuronal loss in AD [54].
Intriguingly, a recent paper published in 2022 demonstrated that the astrocyte-specific isoform of the NKA α subunit, NKAα2, might positively regulate astrocytic-dependent neuroinflammation [44]. Following evidence demonstrated that the expression level of NKAα2 was elevated in human tauopathies and a mouse model of tauopathy. The pharmacological inhibition of NKAα2 robustly suppressed neuroinflammation and reduced brain atrophy. In addition, NKAα2 knockdown in tauopathy mice halted the accumulation of tau pathology [44]. These observations revealed that NKAα2 promoted tauopathy via increased tau uptake in neurons (Figure 2). Taken together, these findings indicate that the roles of different isoforms of NKA in different brain cells may be different and sophisticated. More evidence are still needed to elucidate the relationship between the isoforms of the NKA α subunit and the progression of AD.
This entry is adapted from the peer-reviewed paper 10.3390/cells11244075
References
- Sierra-Fonseca, J.A.; Gosselink, K.L. Tauopathy and neurodegeneration: A role for stress. Neurobiol. Stress 2018, 9, 105–112.
- Kumar, R.; Amruthanjali, T.; Singothu, S.; Singh, S.B.; Bhandari, V. Uncoupling proteins as a therapeutic target for the development of new era drugs against neurodegenerative disorder. Biomed. Pharmacother.=Biomed. Pharmacother. 2022, 147, 112656.
- Ruffini, N.; Klingenberg, S.; Schweiger, S.; Gerber, S. Common Factors in Neurodegeneration: A Meta-Study Revealing Shared Patterns on a Multi-Omics Scale. Cells 2020, 9, 2642.
- Marques-Aleixo, I.; Beleza, J.; Sampaio, A.; Stevanović, J.; Coxito, P.; Gonçalves, I.; Ascensão, A.; Magalhães, J. Preventive and Therapeutic Potential of Physical Exercise in Neurodegenerative Diseases. Antioxid. Redox Signal. 2021, 34, 674–693.
- Teixeira, F.C.; Gutierres, J.M.; Soares, M.S.P.; da Siveira de Mattos, B.; Spohr, L.; do Couto, C.A.T.; Bona, N.P.; Assmann, C.E.; Morsch, V.M.; da Cruz, I.B.M.; et al. Inosine protects against impairment of memory induced by experimental model of Alzheimer disease: A nucleoside with multitarget brain actions. Psychopharmacology 2020, 237, 811–823.
- Del Fabbro, L.; Rossito Goes, A.; Jesse, C.R.; de Gomes, M.G.; Cattelan Souza, L.; Lobo Ladd, F.V.; Lobo Ladd, A.A.B.; Nunes Arantes, R.V.; Reis Simionato, A.; Oliveira, M.S.; et al. Chrysin protects against behavioral, cognitive and neurochemical alterations in a 6-hydroxydopamine model of Parkinson’s disease. Neurosci. Lett. 2019, 706, 158–163.
- Ishrat, T.; Parveen, K.; Hoda, M.N.; Khan, M.B.; Yousuf, S.; Ansari, M.A.; Saleem, S.; Islam, F. Effects of Pycnogenol and vitamin E on cognitive deficits and oxidative damage induced by intracerebroventricular streptozotocin in rats. Behav. Pharmacol. 2009, 20, 567–575.
- Kawamoto, E.M.; Cararo-Lopes, M.M.; Kinoshita, P.F.; Quintas, L.E.M.; Lima, L.S.; Andreotti, D.Z.; Scavone, C. Influence of Nitric Oxide-Cyclic GMP and Oxidative STRESS on Amyloid-β Peptide Induced Decrease of Na,K-ATPase Activity in Rat Hippocampal Slices. J. Membr. Biol. 2021, 254, 463–473.
- Cao, L.; Xiong, S.; Wu, Z.; Ding, L.; Zhou, Y.; Sun, H.; Zhu, M.; Lee, W.T.; Nie, X.; Bian, J.S. Anti-Na(+)/K(+)-ATPase immunotherapy ameliorates α-synuclein pathology through activation of Na(+)/K(+)-ATPase α1-dependent autophagy. Sci. Adv. 2021, 7, eabc5062.
- Raghavan, M.; Fee, D.; Barkhaus, P.E. Generation and propagation of the action potential. Handb. Clin. Neurol. 2019, 160, 3–22.
- Raju, T.N. The Nobel chronicles. 1963: Sir Alan Lloyd Hodgkin (1914–98), Sir Andrew Fielding Huxley (b 1917), and Sir John Carew Eccles (1903–97). Lancet 1999, 354, 263.
- Cui, X.; Xie, Z. Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules 2017, 22, 990.
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535.
- Finel, M.; Haltia, T. The Nobel prize in chemistry for researchers of Na+, K+ ATPase and ATP synthase. Duodecim 1997, 113, 2503–2507.
- Shinoda, T.; Ogawa, H.; Cornelius, F.; Toyoshima, C. Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 2009, 459, 446–450.
- Shull, G.E.; Schwartz, A.; Lingrel, J.B. Amino-acid sequence of the catalytic subunit of the (Na+ + K+)ATPase deduced from a complementary DNA. Nature 1985, 316, 691–695.
- Blanco, G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol. 2005, 25, 292–303.
- Pavone, P.; Pappalardo, X.G.; Incorpora, G.; Falsaperla, R.; Marino, S.D.; Corsello, G.; Parano, E.; Ruggieri, M. Long-term follow-up and novel genotype-phenotype analysis of monozygotic twins with ATP1A3 mutation in Alternating Hemiplegia of Childhood-2. Eur. J. Med. Genet. 2020, 63, 103957.
- Gallanti, A.; Tonelli, A.; Cardin, V.; Bussone, G.; Bresolin, N.; Bassi, M.T. A novel de novo nonsense mutation in ATP1A2 associated with sporadic hemiplegic migraine and epileptic seizures. J. Neurol. Sci. 2008, 273, 123–126.
- Dobretsov, M.; Stimers, J.R. Neuronal function and alpha3 isoform of the Na/K-ATPase. Front. Biosci. A J. Virtual Libr. 2005, 10, 2373–2396.
- Paciorkowski, A.R.; McDaniel, S.S.; Jansen, L.A.; Tully, H.; Tuttle, E.; Ghoneim, D.H.; Tupal, S.; Gunter, S.A.; Vasta, V.; Zhang, Q.; et al. Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 2015, 56, 422–430.
- Dard, R.; Mignot, C.; Durr, A.; Lesca, G.; Sanlaville, D.; Roze, E.; Mochel, F. Relapsing encephalopathy with cerebellar ataxia related to an ATP1A3 mutation. Dev. Med. Child Neurol. 2015, 57, 1183–1186.
- Demos, M.K.; van Karnebeek, C.D.; Ross, C.J.; Adam, S.; Shen, Y.; Zhan, S.H.; Shyr, C.; Horvath, G.; Suri, M.; Fryer, A.; et al. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J. Rare Dis. 2014, 9, 15.
- Sanchez, G.; Nguyen, A.N.; Timmerberg, B.; Tash, J.S.; Blanco, G. The Na,K-ATPase alpha4 isoform from humans has distinct enzymatic properties and is important for sperm motility. Mol. Hum. Reprod. 2006, 12, 565–576.
- Ohnishi, T.; Yanazawa, M.; Sasahara, T.; Kitamura, Y.; Hiroaki, H.; Fukazawa, Y.; Kii, I.; Nishiyama, T.; Kakita, A.; Takeda, H.; et al. Na, K-ATPase α3 is a death target of Alzheimer patient amyloid-β assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E4465–E4474.
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Léna, C.; et al. α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 2015, 34, 2408–2423.
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. 1998, 275, F633–F650.
- Geering, K. The functional role of beta subunits in oligomeric P-type ATPases. J. Bioenerg. Biomembr. 2001, 33, 425–438.
- Gloor, S.; Antonicek, H.; Sweadner, K.J.; Pagliusi, S.; Frank, R.; Moos, M.; Schachner, M. The adhesion molecule on glia (AMOG) is a homologue of the beta subunit of the Na,K-ATPase. J. Cell Biol. 1990, 110, 165–174.
- Tokhtaeva, E.; Sachs, G.; Sun, H.; Dada, L.A.; Sznajder, J.I.; Vagin, O. Identification of the amino acid region involved in the intercellular interaction between the β1 subunits of Na+/K+-ATPase. J. Cell Sci. 2012, 125, 1605–1616.
- Ackermann, U.; Geering, K. Mutual dependence of Na,K-ATPase alpha- and beta-subunits for correct posttranslational processing and intracellular transport. FEBS Lett. 1990, 269, 105–108.
- Garty, H.; Karlish, S.J. Role of FXYD proteins in ion transport. Annu. Rev. Physiol. 2006, 68, 431–459.
- Geering, K. FXYD proteins: New regulators of Na-K-ATPase. Am. J. Physiol. Ren. Physiol. 2006, 290, F241–F250.
- Geering, K.; Béguin, P.; Garty, H.; Karlish, S.; Füzesi, M.; Horisberger, J.D.; Crambert, G. FXYD proteins: New tissue- and isoform-specific regulators of Na,K-ATPase. Ann. N. Y. Acad. Sci. 2003, 986, 388–394.
- Waxman, S.G.; Ritchie, J.M. Molecular dissection of the myelinated axon. Ann. Neurol. 1993, 33, 121–136.
- Cardone, R.A.; Alfarouk, K.O.; Elliott, R.L.; Alqahtani, S.S.; Ahmed, S.B.M.; Aljarbou, A.N.; Greco, M.R.; Cannone, S.; Reshkin, S.J. The Role of Sodium Hydrogen Exchanger 1 in Dysregulation of Proton Dynamics and Reprogramming of Cancer Metabolism as a Sequela. Int. J. Mol. Sci. 2019, 20, 3694.
- Blaustein, M.P.; Hamlyn, J.M. Ouabain, endogenous ouabain and ouabain-like factors: The Na(+) pump/ouabain receptor, its linkage to NCX, and its myriad functions. Cell Calcium 2020, 86, 102159.
- Liu, L.; Ivanov, A.V.; Gable, M.E.; Jolivel, F.; Morrill, G.A.; Askari, A. Comparative properties of caveolar and noncaveolar preparations of kidney Na+/K+-ATPase. Biochemistry 2011, 50, 8664–8673.
- Shimizu, H.; Watanabe, E.; Hiyama, T.Y.; Nagakura, A.; Fujikawa, A.; Okado, H.; Yanagawa, Y.; Obata, K.; Noda, M. Glial Nax channels control lactate signaling to neurons for brain sensing. Neuron 2007, 54, 59–72.
- Petrushanko, I.Y.; Mitkevich, V.A.; Anashkina, A.A.; Adzhubei, A.A.; Burnysheva, K.M.; Lakunina, V.A.; Kamanina, Y.V.; Dergousova, E.A.; Lopina, O.D.; Ogunshola, O.O.; et al. Direct interaction of beta-amyloid with Na,K-ATPase as a putative regulator of the enzyme function. Sci. Rep. 2016, 6, 27738.
- Alzheimer, A. Uber eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. Psych.-Gerichtl. Med. 1907, 46, 146–148.
- Maggiore, A.; Casale, A.M.; Toscanelli, W.; Cappucci, U.; Rotili, D.; Grieco, M.; Gagné, J.P.; Poirier, G.G.; d’Erme, M.; Piacentini, L. Neuroprotective Effects of PARP Inhibitors in Drosophila Models of Alzheimer’s Disease. Cells 2022, 11, 1284.
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 2022, 214, 102270.
- Santiago, J.A.; Potashkin, J.A. The Impact of Disease Comorbidities in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 631770.
- Parker, K.; Rhee, Y. Alzheimer’s Disease Warning Signs: Gender and Education Influence Modifiable Risk Factors—A Pilot Survey Study. J. Am. Coll. Nutr. 2021, 40, 583–588.
- Xiong, J.; Kang, S.S.; Wang, Z.; Liu, X.; Kuo, T.C.; Korkmaz, F.; Padilla, A.; Miyashita, S.; Chan, P.; Zhang, Z.; et al. FSH blockade improves cognition in mice with Alzheimer’s disease. Nature 2022, 603, 470–476.
- Hattori, N.; Kitagawa, K.; Higashida, T.; Yagyu, K.; Shimohama, S.; Wataya, T.; Perry, G.; Smith, M.A.; Inagaki, C. CI-ATPase and Na+/K(+)-ATPase activities in Alzheimer’s disease brains. Neurosci. Lett. 1998, 254, 141–144.
- Liguri, G.; Taddei, N.; Nassi, P.; Latorraca, S.; Nediani, C.; Sorbi, S. Changes in Na+,K(+)-ATPase, Ca2(+)-ATPase and some soluble enzymes related to energy metabolism in brains of patients with Alzheimer’s disease. Neurosci. Lett. 1990, 112, 338–342.
- Sharoar, M.G.; Palko, S.; Ge, Y.; Saido, T.C.; Yan, R. Accumulation of saposin in dystrophic neurites is linked to impaired lysosomal functions in Alzheimer’s disease brains. Mol. Neurodegener. 2021, 16, 45.
- Tian, J.; Shi, J.; Zhang, L.; Yin, J.; Hu, Q.; Xu, Y.; Sheng, S.; Wang, P.; Ren, Y.; Wang, R.; et al. GEPT extract reduces Abeta deposition by regulating the balance between production and degradation of Abeta in APPV717I transgenic mice. Curr. Alzheimer Res. 2009, 6, 118–131.
- Joshi, Y.B.; Giannopoulos, P.F.; Praticò, D. The 12/15-lipoxygenase as an emerging therapeutic target for Alzheimer’s disease. Trends Pharmacol. Sci. 2015, 36, 181–186.
- Dorard, E.; Chasseigneaux, S.; Gorisse-Hussonnois, L.; Broussard, C.; Pillot, T.; Allinquant, B. Soluble Amyloid Precursor Protein Alpha Interacts with alpha3-Na, K-ATPAse to Induce Axonal Outgrowth but Not Neuroprotection: Evidence for Distinct Mechanisms Underlying these Properties. Mol. Neurobiol. 2018, 55, 5594–5610.
- Yang, Y.; Zhang, J.; Yang, X.; Li, Z.; Wang, J.; Lu, C.; Nan, A.; Zou, Y. Dysregulated APP expression and α-secretase processing of APP is involved in manganese-induced cognitive impairment. Ecotoxicol. Environ. Saf. 2021, 220, 112365.
- Ahmad, R.; Khan, A.; Lee, H.J.; Ur Rehman, I.; Khan, I.; Alam, S.I.; Kim, M.O. Lupeol, a Plant-Derived Triterpenoid, Protects Mice Brains against Aβ-Induced Oxidative Stress and Neurodegeneration. Biomedicines 2020, 8, 380.
- Lakunina, V.A.; Petrushanko, I.Y.; Burnysheva, K.M.; Mitkevich, V.A.; Makarov, A.A. Alzheimer’s disease Aβ(42) peptide induces an increase in Na,K-ATPase glutathionylation. Dokl. Biochem. Biophys. 2017, 473, 114–117.
- Sasahara, T.; Satomura, K.; Tada, M.; Kakita, A.; Hoshi, M. Alzheimer’s Aβ assembly binds sodium pump and blocks endothelial NOS activity via ROS-PKC pathway in brain vascular endothelial cells. iScience 2021, 24, 102936.
- Tian, Y.; Qi, Y.; Cai, H.; Xu, M.; Zhang, Y. Senegenin alleviates Aβ(1-42) induced cell damage through triggering mitophagy. J. Ethnopharmacol. 2022, 295, 115409.
- Mitkevich, V.A.; Petrushanko, I.Y.; Poluektov, Y.M.; Burnysheva, K.M.; Lakunina, V.A.; Anashkina, A.A.; ef-17 Makarov, A.A. Basal Glutathionylation of Na,K-ATPase α-Subunit Depends on Redox Status of Cells during the Enzyme Biosynthesis. Oxidative Med. Cell. Longev. 2016, 2016, 9092328.
- da Silva, F.D.; Pinz, M.P.; de Oliveira, R.L.; Rodrigues, K.C.; Ianiski, F.R.; Bassaco, M.M.; Silveira, C.C.; Jesse, C.R.; Roman, S.S.; Wilhelm, E.A.; et al. Organosulfur compound protects against memory decline induced by scopolamine through modulation of oxidative stress and Na(+)/K(+) ATPase activity in mice. Metab. Brain Dis. 2017, 32, 1819–1828.
- Mark, R.J.; Keller, J.N.; Kruman, I.; Mattson, M.P. Basic FGF attenuates amyloid beta-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na+/K+-ATPase activity in hippocampal neurons. Brain Res. 1997, 756, 205–214.
- Xin, S.H.; Tan, L.; Cao, X.; Yu, J.T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox. Res. 2018, 34, 733–748.
This entry is offline, you can click here to edit this entry!