Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Hepatitis B virus (HBV) and hepatitis delta virus (HDV) are highly prevalent viruses estimated to infect approximately 300 million people and 12–72 million people worldwide, respectively. HDV requires the HBV envelope to establish a successful infection. Concurrent infection with HBV and HDV can result in more severe disease outcomes than infection with HBV alone. These viruses can cause significant hepatic disease, including cirrhosis, fulminant hepatitis, and hepatocellular carcinoma, and represent a significant cause of global mortality.
- HBV
- HDV
- HBsAg
- HDAg
- immune response
1. Introduction
Hepatitis B virus (HBV) is a highly prevalent virus worldwide. Indeed, it was estimated that 248 million individuals were hepatitis B surface antigen (HBsAg) positive as of 2010 [1]. HBsAg is a HBV protein that serves as a marker of HBV infection [2]. According to World Health Organization (WHO) data from 2019, the case number is rising, with 296 million individuals living with chronic hepatitis B infection and 1.5 million new infections each year. The WHO data further shows that almost 1 million persons die annually from complications of chronic hepatitis B infection [3]. The most common mode of HBV transmission is through exposure to infected blood or bodily fluids [4]. HBV is often spread perinatally at birth, during sexual contact, or through recreational injection drug use [4]. Spread in the healthcare setting is associated with exposure to contaminated blood or blood products [4], such as needle sticks [5] or transfusions [6]. While most infected adults clear the virus, 5–10% develop a chronic HBV infection and subsequent hepatic disease that can be quite severe. Examples of serious outcomes include acute hepatitis/fulminant hepatic failure, cirrhosis, and hepatocellular carcinoma [7]. Extrahepatic manifestations, such as polyarteritis nodosa, arthralgia, neuritis, vasculitis, and glomerulonephritis, may also complicate HBV infection [8][9][10]. Many of these adverse effects may be secondary to immune-mediated damage rather than the direct cytopathic effects of the virus itself [11][12][13][14]. HBV-specific effector CD8+ cells induce hepatocyte apoptosis in infected individuals. If these apoptotic cells are not quickly cleared by Kupffer cells, the apoptotic hepatocytes can release damage-associated molecular patterns that result in the recruitment of neutrophils and a subsequent damaging immune response [14]. However, it is worth noting that there is evidence suggesting HBV-mediated damage may play a role as well [15][16]. HBV and associated diseases represent a significant cause of worldwide mortality. In fact, it was estimated that HBV caused 39% of the approximately 820,000 deaths secondary to hepatic cancer and 29% of the 1.32 million deaths secondary to cirrhosis worldwide in 2017 [17].
HBV has been classified into at least 10 genotypes labeled A-J [18][19], as well as several subtypes. These classifications differ in several factors such as global distribution, gene expression levels, and clinical course [18][19][20][21]. For example, genotype A was associated with prolonged HBsAg elevation and development of chronic HBV following acute hepatitis infection in Japanese adults [22], and genotype C has been associated with hepatocellular carcinoma (HCC) [23]. The genotype also has implications for response to treatment. For example, genotypes A and B were shown to be more likely to seroconvert than genotype D following treatment with pegylated interferon (PEG-IFN) in hepatitis E antigen (HBeAg)-positive patients [24]. Following cessation of tenofovir disoproxil fumarate or entecavir in HBeAg- negative patients, those with genotype B are more likely to relapse and require retreatment than genotype C [25].
Hepatitis D virus (HDV) is considered a satellite virus because it requires HBV surface proteins to generate mature virion particles [26]. As a result, HDV can establish either co-infection or superinfection with HBV, but cannot package infectious virions or spread in the absence of HBV [26]. It was previously demonstrated that enveloped viruses distinct from HBV can form infectious delta virus particles [27], but there is currently no firm evidence for the clinical significance of these observations gleaned from cell cultures and mouse models. Coinfected individuals may develop an acute hepatitis but are typically able to clear both viruses. HDV superinfection of a chronically infected HBV carrier is more likely to result in chronic HDV infection with its concomitant comorbidities. Development of a chronic HDV superinfection can exacerbate hepatic injury caused by HBV [26][28], and both coinfection and superinfection have been shown to result in more severe outcomes than HBV infection alone, including fulminant hepatitis, HCC, and chronic hepatitis [26][29].
While not quite as prevalent as HBV, a study published in September 2020 estimated that 12 million individuals worldwide are seropositive for anti-HDV antibodies [30]. Notably, there has been some variance regarding estimates of HDV prevalence, with a separate study placing the number of infected individuals as high as 62–72 million [31], while another study published in May 2020 estimated that there are between 48–60 million HDV infections worldwide [29]. Modes of HDV transmission include exposure to contaminated blood, high risk sexual behavior, and injection drug use. [32]. The risk of vertical HDV transmission from mother to child is relatively low [33]. Chronic hepatitis D is considered the most severe of all hepatitis infections [34] and is associated with increased risks of hepatocellular carcinoma, cirrhosis, liver decompensation, and mortality [35]. Like HBV, HDV-associated disease is considered to be largely immune-mediated in nature [36].
HBV and HDV Cell Tropism
While HBV primarily infects hepatocytes in vivo, it has been shown to infect non-hepatic cells in vivo as well, including but not limited to endothelial cells, hematopoietic precursors, neuronal cells, and monocytes/macrophages [37]. The primary cell lines used for in vitro experiments are hepatic cell lines including primary human hepatocytes, primary tupaia hepatocytes, and the hepatic cancer lines HepaRG, Huh7, and HepG2. These cell lines also support in vitro experiments involving HDV (reviewed in [38]). The discovery of sodium taurocholate cotransporting polypeptide (NTCP), a multiple transmembrane transporter expressed in the human liver, as a key entry receptor for both HBV and HDV [39] has resulted in engineered cell lines. These cell lines, such as HepG2-NTCP and Huh7-NTCP, are susceptible to infection by both viruses [38][40]. Only some primates and tree shrews are susceptible to human HBV infection, limiting in vivo experimentation. However, models of HBV infection based on related viruses exist in woodchucks, ground squirrels, and ducks [41]. Organisms naturally susceptible to HDV infection are similarly few in number and include chimpanzees and tree shrews [42]. Human liver chimeric mice supporting both HBV and HDV infection have also been developed [42][43], and mice models carrying HBV and HDV infection have been created through hydrodynamic delivery [44][45][46]. Numerous additional approaches have been taken to model HBV and HDV infection in animal models, which have reviewed recently [42][47].
2. Virion Structure and Viral Life Cycle of HBV and HDV
HBV is a member of the Hepadnaviridae family and belongs to the genus Orthohepadnavirus. The virion is approximately 42 nm in diameter. A host-derived membrane that contains small (S-HBsAg), middle (M-HBsAg), and large (L-HBsAg) hepatitis B surface antigens [48][49] surrounds an icosahedral capsid comprised of the core antigen [50]. HBV’s partially double-stranded, relaxed circular DNA (rcDNA) genome, which is approximately 3.2 kb in length, includes a lesion bearing a plus and minus strand. The HBV polymerase is covalently attached via a 10 nucleotide DNA flap to the minus strand. The plus strand contains a gap and an RNA primer which is a remnant of the reverse transcription reaction [51][52]. Figure 1a below demonstrates the structure of HBV and HDV, while Figure 1b illustrates their genomic structures.
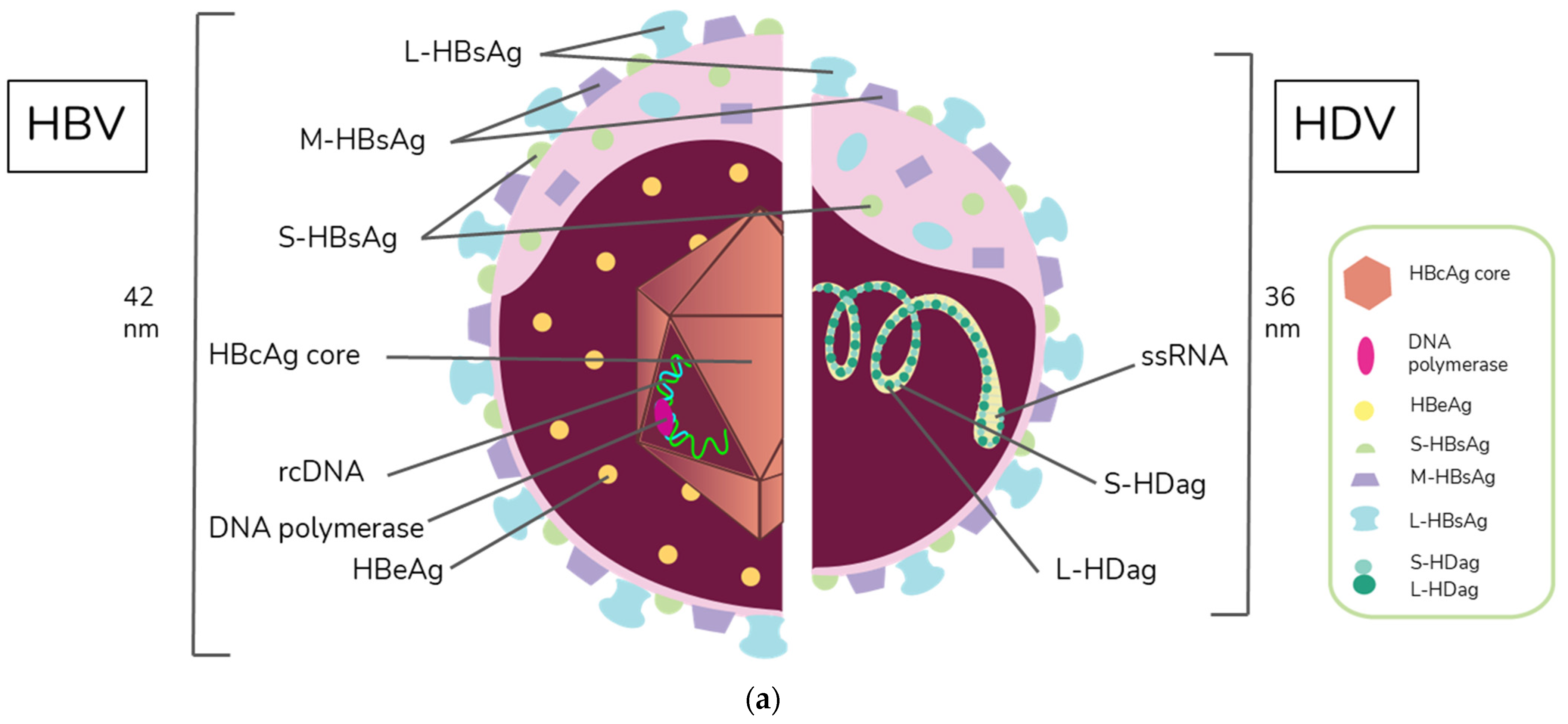
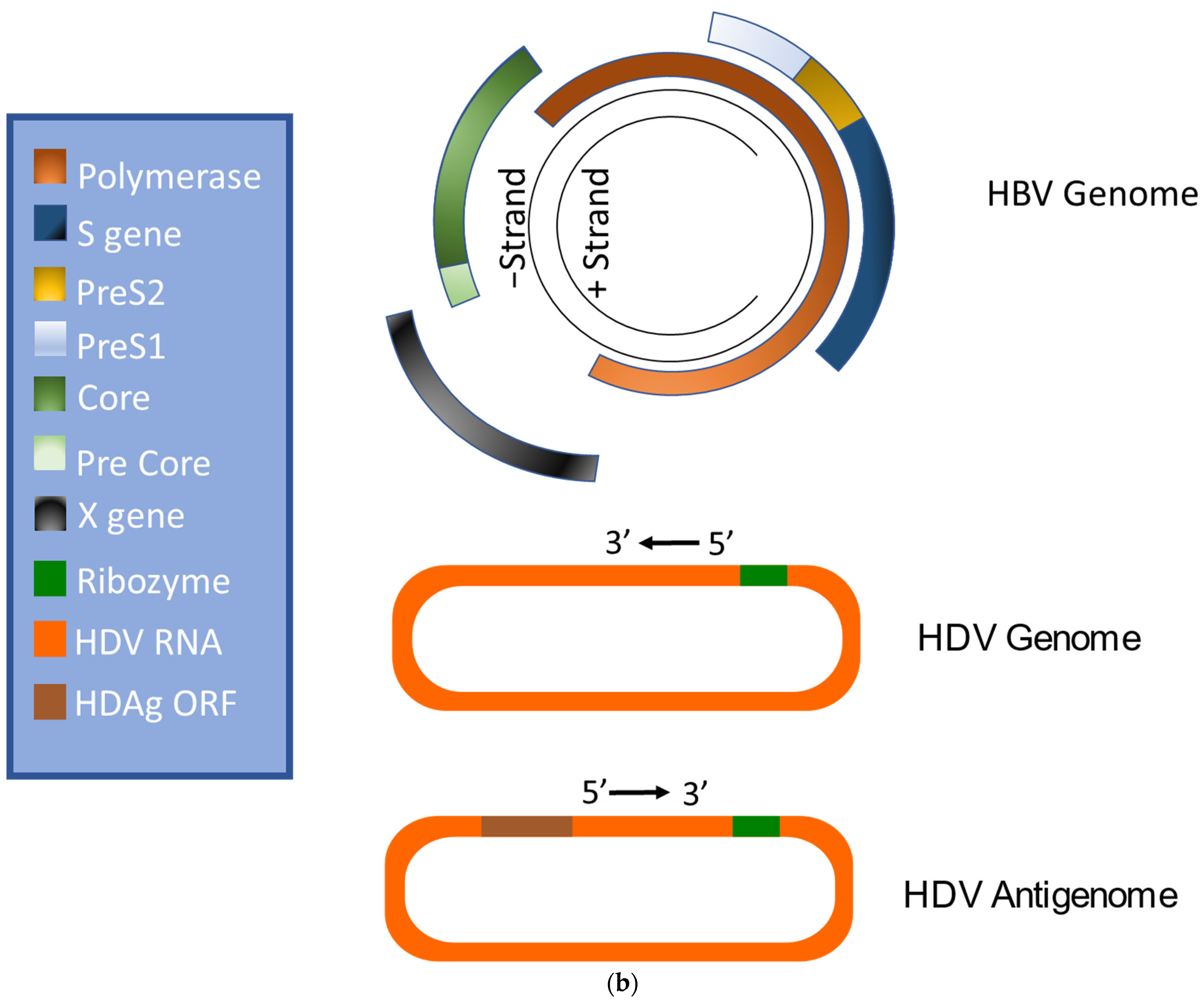
Figure 1. Virion and genome structures of hepatitis B virus (HBV) and hepatitis D virus (HDV). (a) Virion Structures of Hepatitis B Virus (HBV) and Hepatitis Delta Virus (HDV). Both HBV and HDV are enveloped in HBV surface proteins designated as small (S-HBsAg), middle (M-HBsAg), and large (L-HBsAg). The HBV relaxed-circular DNA (rcDNA) is partially double-stranded, with a full minus strand and incomplete plus strand, and a surrounding icosahedral capsid comprised of the core antigen. HBeAg proteins are present between the envelope and capsid. The HDV virion contains a single-stranded RNA genome that is complexed with small and large delta antigens (S-HDAg and L-HDAg, respectively). (b) Genomic Structure of HBV and HDV. HBV is a partially dsDNA virus composed of four distinct open reading frames (ORFs): polymerase, S, core, and X. There is significant overlap between the different ORFs. The RNA of both the single-stranded RNA genome and the antigenome contain self-cleaving ribozymes [53]. Unedited antigenome ultimately codes for S-HDAg. The cellular enzyme adenosine deaminase acting on RNA 1 (ADAR1) ADAR1 can modify the antigenome to generate the genetic code for L-HDAg [54].
The HBV entry process into hepatocytes can be subdivided into several steps. In the first step, the virus attaches to the surface of the host cell by binding to heparan sulfate proteoglycans (HSPGs) such as glypican 5 [55]. Following this initial attachment, HBV is thought to bind to its dedicated receptor–NTCP [39]. Notably, Perez-Vargas et al. recently showed that neither HSPGs nor NTCP were required to induce viral fusion, but that there was a key fusion peptide in the preS1 domain of L-HBsAg [56]. The authors propose that regulation of cellular cross strand disulfide bonds by the cellular protein disulfide isomerase ERp57 positions the preS1 fusion peptide to facilitate fusion between the viral and endosomal membrane [56]. HBV is believed to enter the cell via the clathrin-mediated endocytosis pathway. The viral capsid is subsequently directed to the nucleus [57], where its rcDNA is modified by cellular factors including proliferating cell nuclear antigen, replication factor C complex, flap endonuclease 1, DNA polymerase δ, and DNA ligase 1 [58] and is eventually transformed into cccDNA. cccDNA is then used as a template for transcription [57]. It is believed that this cccDNA is directly responsible for establishing chronicity [59].
Four different lengths of RNAs are then transcribed, pre-C/C, pre-S, S, and X mRNA. These transcripts are 3.5 kb, 2.4 kb, 2.1 kb, and 0.7 kb in length, respectively [51]. Pregenomic RNA (PgRNA), which is the RNA sequence produced following transcription from cccDNA [60], is then transported to the cytoplasm. Once there, the pgRNA is encapsidated with HBV polymerase, and rcDNA is created via reverse transcription [61]. Virions containing rcDNA are then enveloped, and the now infectious virions are secreted in multivesicular bodies (MVBs) [61][62].
HBV encodes the viral antigens HBsAg from pre-S and S mRNA and HBx, which is involved in infection and replication, from mRNA to the X gene [7][51]. HBsAg is the viral envelope protein and is encoded by the S gene [7][51]. It can be expressed as S-HBsAg, M-HBsAg, or L-HBsAg [50]. HBV also encodes HBeAg and core antigen (HBcAg) [7][51]. The C gene codes for HBcAg and a precore protein. The precore protein is then processed by the golgi and endoplasmic reticulum and secreted as HBeAg [63].
HDV is the only member of the Deltavirus genus and measures approximately 36 nm in diameter [64]. The core of the HDV virion is a single-stranded RNA molecule complexed with both the small and large forms of hepatitis D antigen (HDAg), the only protein encoded by HDV [64]. This core is surrounded by an envelope comprised of the HBV envelope proteins S-HBsAg, M-HBsAg, and L-HBsAg. While M-HBsAg is thought to be nonessential for HDV infectivity, both L-HBsAg and S-HBsAg are required [54].
Like HBV, HDV attaches to the host’s surface HSPGs [65] and uses NTCP for cell entry [39]. The ribonucleoprotein core travels to the hepatocyte nucleus for replication. There, the viral RNA genome replicates, generating approximately 300,000 new viral copies as well as a smaller number of complementary RNA antigenome [64], which is a replication intermediate rather than mRNA [66]. This is edited during the replication step by the cellular enzyme adenosine deaminase acting on RNA (ADAR) [67][68] to produce a second distinct antigenome strand [54] that contains the open reading frame (ORF) for HDAg [69]. Replication is accomplished in a rolling-circle fashion, and autocatalytic ribozymes present in both the genome and antigenome function to cleave the strands into linear RNA strands that are ligated into the circular antigenome RNA [70]. As HDV does not have its own RNA polymerases, replication is presumably accomplished by hijacking host enzymes [64][71]. There is supporting evidence that RNA polymerase II is used to transcribe HDV mRNA [72][73]. Both RNA polymerase I and III are known to bind to HDV RNA [74]. While previous experiments have linked RNA polymerase I with the antigenome, the function of RNA polymerase III remains undiscovered [71][74][75]. Figure 2 illustrates the viral cycles of HBV and HDV.
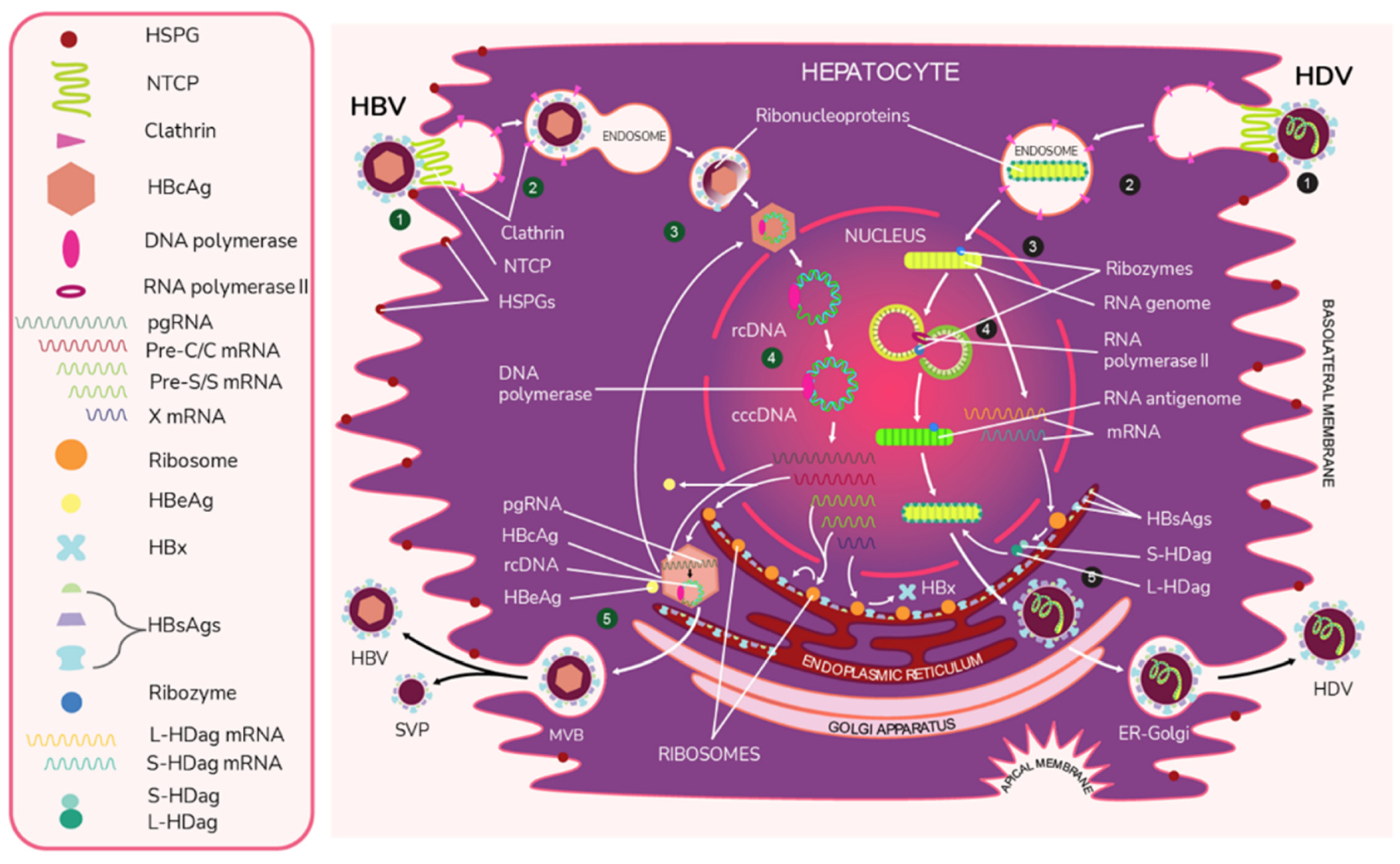
Figure 2. The viral life cycles of HBV and HDV. HBV (green numbers): (1) HBV attaches to the hepatocyte by first binding to host heparan sulfate proteoglycans (HSPGs) and then binding to the liver cell entry receptor sodium taurocholate cotransporting polypeptide (NTCP) with the pre-S1 domain of L-HBsAg. (2) HBV enters the cell via the clathrin-mediated endocytosis pathway through clathrin protein interactions with L-HbsAg on the HBV membrane. (3) The viral envelope fuses to the early endosome, and the core protein of the exposed nucleocapsid binds to the host nucleus and releases its viral DNA and DNA polymerase. * (4) The virus begins replication by first completing its partially double-stranded relaxed circular (rcDNA) using DNA polymerase to create full dsDNA in the form of covalently closed circular DNA (cccDNA). The DNA is then transcribed by host RNA polymerase II into pregenomic RNA (pgRNA) and 4 mRNAs. The 4 mRNAs are translated by host ribozymes. The “Pre-C/C” mRNA is translated into HBcAg core and HBeAg, the “Pre-S” and “S” mRNAs into HBV surface proteins, and “X” mRNA into HBx protein, which is involved in infection and replication. The pgRNA is transported to the cytoplasm, where it is encapsidated by the HBcAg core. Inside this preliminary nucleocapsid, HBV polymerase reverse transcribes pgRNA into negative-sense DNA, which undergoes synthesis into partially double-stranded rcDNA. The completed nucleocapsid either travels back to the nucleus to create additional virions or buds from the ER membrane, acquiring an envelope that is embedded with HBsAg. (5) Along with the infectious virions, empty subviral particles (SVPs) also bud out from the ER. The exiting viral and subviral particles pass through the Golgi apparatus into multivesicular bodies (MVBs), which then bind to the hepatocyte envelope and are released via exocytosis. HDV (black numbers): (1) HDV attaches to the host cell through low-specificity binding to HSPGs and high-specificity binding to NTCP with the viral L-HBsAg pre-S1 domain. (2) The virus enters the cell. (3) The viral envelope is uncoated and the ribonucleoprotein targets the hepatocyte nucleus. * (4) In a rolling circle manner, the viral RNA genome is transcribed into the antigenome, and the antigenome serves as a template for new genome transcripts, both of which self-cleave and ligate to reform the circular RNA. The genome also produces S-HDAg and L-HDAg mRNA from the same open reading frame. This process produces approximately 300,000 genome and a smaller number of antigenome molecules. (5) The translated protein products re-enter the nucleus to complete replication and particle assembly. Fully assembled HDV virions pass through the ER-Golgi complex and exit the host cell. * The exact uncoating mechanisms of the envelope and nucleocapsid are not fully understood.
HDAg is ultimately translated into two isoforms: small HDAg (S-HDAg) and large HDAg (L-HDAg). This process begins during replication, with the unedited antigenome strand producing mRNA that is translated into S-HDAg and another strand that is edited by ADAR1 to produce L-HDAg mRNA [54][76][77]. Specifically, ADAR1 alters a UAG (stop) codon to UGG (tryptophan), which allows for the expression of the larger L-HDAg [54][76][78]. S-HDAg functions to upregulate RNA production and is thought to recruit the host RNA polymerase for replication. This is because it binds to RNA polymerase II [64], possibly by way of chromatin remodeling complex recruitment. It has also been shown to interact with polymerase I. S-HDAg is required for replication [54]. In contrast, L-HDAg undergoes the post-transcriptional modification prenylation [54][79], which inhibits further RNA accumulation and contributes to the assembly of new viral particles [54].
Ribonucleoprotein assembly occurs in the hepatocyte nucleus. Assuming the viral particle can obtain the necessary HBV envelope proteins, the infectious delta virions are thought to exit the cell through the golgi [27][54].
3. Natural Course of HBV and HDV Infection
According to guidelines established by the American Association for the Study of Liver Diseases, there are four phases of chronic hepatitis B infection [80], All four phases are defined by the presence of HBsAg for ≥6 months. In the first phase of chronic HBV infection, serum HBV levels range from undetectable to several billion IU/mL. Patients in this phase are subdivided into either HBeAg positive or negative. HBV-DNA levels are often > 20,000 IU/mL in HBeAg-positive chronic HBV, while lower values of 2000–20,000 IU/mL are typically seen in HBeAg-negative chronic HBV patients. ALT and AST levels can be either normal or elevated, and liver biopsy demonstrates chronic hepatitis with variable necroinflammation and/or fibrosis [80]. Patients in the second phase, known as the Immune-Tolerant (IT) phase, are HBeAg positive with elevated HBV DNA levels often >1 million IU/mL. ALT/AST levels are normal or slightly elevated. Liver biopsy performed during this stage demonstrates minimal inflammation with no fibrosis [80]. Patients in the third phase, known as Immune-Active (IA) chronic HBV, have differing HBV DNA levels based on their HBeAg status. HBeAg-positive patients have serum HBV DNA levels > 20,000 IU/mL, while HBeAg-negative patients have lower levels of at least 20,000 IU/mL. ALT/AST levels at this stage are sporadically or persistently elevated consistent with host immune system-mediated hepatic injury [80]. Liver biopsy or noninvasive testing at this stage reveal chronic hepatitis and moderate to severe necroinflammation. Fibrosis may or may not be present [80]. The fourth and final phase is termed inactive chronic HBV. Patients in this phase are HBeAg negative and anti-HBe positive. Serum HBV DNA levels are typically <2000 IU/mL, and ALT/AST levels are firmly within normal ranges. Liver biopsy at this stage demonstrates varying degrees of fibrosis without significant necroinflammation [80]. An important consideration is that chronic HBV is dynamic; patients may transition between phases in either direction [81].
The risk of developing a chronic infection is not uniform; instead, it is inversely proportional to age [82][83] For example, the risk of HBV transmission from mother to baby is quite high [84]. For vertically infected infants whose mother is positive for both HBsAg and HBeAg, the risk of progression to chronic HBV infection is between 85–90% in the absence of passive-active prophylaxis with hepatitis B immune globulin and the HBV vaccine [85]. These rates decrease for children younger than 5 to between 20–50% and are the lowest for older children and adults, with between 5–10% progressing to chronic HBV [82].
There are two distinct patterns of infection for HDV: co-infection, which involves simultaneous infection with HBV and HDV, and superinfection, which involves the infection of an HBsAg-positive patient with HDV [86]. The pattern of infections typically influences the clinical outcome. Coinfection typically causes a more severe hepatitis with a greater incidence of acute liver failure than acute HBV infection alone. It also may lead to biphasic peaks in AST/ALT. [87]. However, patients are typically able to clear coinfections without developing chronic HBV/HDV infection. On the other hand, superinfection typically results in severe acute hepatitis with subsequent progression to chronic hepatitis in over 90% of cases [88]. Because of the differences in prognosis and treatment, discriminating between acute co-infection and superinfection in these patients is critical [89].
While the pathogenesis of HDV-mediated liver damage is not well understood, the pathogenesis of HDV-related liver disease can be attributed to several factors including HDV genotype, HBV genotype, and the host immune response [90]. The clinical characteristics of HDV infection are not easily distinguished from other causes of hepatitis and must be differentiated using diagnostic tests [91]. HDV is diagnosed by screening for HDAg-specific antibodies, namely anti-HDV IgM and IgG, in HBsAg-positive individuals. Patients with the anti-HDV reagent should be screened for serum HDV RNA to differentiate between active infection (HDV RNA positive) and a decreasing serological scar (HDV RNA negative) [89].
A study conducted by Genesca et al. assessed the effects of HDV co- and superinfection on HBV replication [92]. Levels of HBV DNA were similar in patients with acute HBV infection, HBV/HDV coinfection, and HBV DNA-positive patients with HBV/HDV superinfection [92]. Of the 37 patients with HBV/HDV coinfection, one died due to fulminant hepatitis and two developed chronic infections. The remaining 34 cleared the infection within three months. One of the chronic patients developed a serological pattern featuring active infection markers of both HBV and HDV. The other patient became HBeAg and HBV DNA-negative within 4 weeks, although HBsAg and HDV antibodies were detectable for longer than two years [92]. In the superinfection group, only 25% of HBsAg carriers (6/24) had detectable HBV DNA while acutely superinfected. Chronic liver disease was documented in all 14 long-term follow up patients with superinfection. Intriguingly, HBV DNA could not be detected in 10 of these patients, although there was evidence for active HDV infection. The data indicated at least a transient inhibition of HBV replication in the presence of HDV superinfection in the four patients with initially detectable HBV DNA. In two of these patients, the time at which HBV DNA became detectable correlated with the time at which serologic markers for HDV replication became undetectable [92]. The authors concluded that patients with inactive HBV infection and HDV superinfection are more likely to have ongoing HDV replication than those with an active HBV infection at the time of superinfection [92].
3.1. HDV RNA and HBV DNA Interplay during Chronic Infection
There is limited information on the natural history of the viral kinetics of patients with chronic HBV and HDV infections. Giersch et al. [93] reported that while HDV often appears to be dominant in co-infected patients, the interplay between HDV RNA and HBV DNA can be “highly dynamic” over the course of infection. Previously, Schaper et al. [94] and later Braga et al. [95] suggested three kinetic profiles (patterns): HDV dominant, HBV dominant, and HDV/HBV equivalent. During anti-HDV treatment with lonafarnib, Mhlanga et al. [96] demonstrate a rise in HBV DNA as HDV RNA is suppressed by treatment, resulting in HBV dominance further confirming the range of kinetic profiles possible. What follows is a description of the kinetic profiles of the viruses during chronic infection within the individual and aggregate data (Table 1). Further studies are needed for understanding the natural interplay of HBV and HDV.
Table 1. HDV RNA and HBV DNA profiles during chronic infection.
| Study|Viral Profile | HDV Dominant |
HBV Dominant |
HDV/HBV Equivalent * | HBeAg (+/−) | HDV RNA Assay ** | Reference |
|---|---|---|---|---|---|---|
| Individual Patient Data | ||||||
| Yurdaydin et al., 2018 | 17/20 | 0/20 | 3/20 | 3 (+) 12 (−) 5 (N/A) |
In-house PCR. (LLoQ) 70 IU/mL and (LLoD) 50 IU/mL |
[97] |
| Shekhtman et al., 2020 | 12/12 | 0/12 | 0/12 | 12 (−) | Robogene MK I LLoQ 3.26 log U/mL |
[98] |
| Schaper et al., 2010 | 13/25 | 6/25 | 6/25 | 5 (+) 20 (−) |
rt-RT-PCR in the LightCyclerTM system |
[94] |
| Guedj et al., 2014 | 11/12 | 0/12 | 1/12 | 9 (−) 3 (+) |
real-time PCR (external) (LLOQ): 100 (GE/mL) |
[99] |
| Koh et al., 2015 | 8/9 | 0/9 | 1/9 | 9 (−) | qRT-PCR LLoD 70 IU/mL |
[100] |
| Patient Data (Cohort Mean) | ||||||
| Castelnau et al., 2006 | 14 | NA | NA | All negative | RT-PCR 100 copies/mL |
[101] |
| Manesis et al., 2007 | 53 | NA | NA | All negative | PCR TaqMan Universal 100 copies/mL of serum and the linearity of quantification ranged from 103 to 109 copies/mL. |
[102] |
| Zachou et al., 2009 *** | 73 | NA | NA | (+) 12 (−) 68 |
RT-PCR | [103] |
| Wedemeyer et al., 2011 | 90 | NA | NA | 14 (+) 76 (−) |
TaqMan One Step PCR Master Mix LLOD of HDV RNA was 120 copies/mL of serum |
[104] |
| Braga et al., 2014 (64 patients–only % given) |
56.3% | 3.1% | 40.6% | All non-reactive | qRT-PCR Sensitivity not listed. |
[95] |
| Koh et al., 2015 | 14 | NA | NA | All negative | qRT-PCR LLoD 70 IU/mL |
[100] |
| Bogomolov et al., 2016 | 8 | 0 | 16 | All Negative | Amplisense HDV-FL During treatment: qPCR: limit of detection (LOD) 15 copies/ml |
[105] |
| Yurdaydin et al., 2007 | 39 | 0 | 0 | (+) 2 (−) 37 |
RT-PCR Sensitivity: 100,000 copies/mL |
[106] |
3.2. Histopathology
Most forms of viral hepatitis, including HBV and HDV, cause necrosis and inflammation of hepatocytes in a similar and indistinguishable manner from one another [107]. In acute disease, there is widespread focalnecrosis, swelling and apoptosis of hepatocytes, and portal tract/intrasinusoidal inflammation. The inflammatory infiltrate is primarily composed of lymphocytes, although plasma cells, neutrophils, and eosinophils may also be seen [107]. Acute hepatitis pathology includes the presence of several histological features. Numerous apoptotic bodies (also called acidophil bodies or Councilman bodies) are seen, as is activation of Kupffer cells (KC, the tissue-resident macrophages of the liver [108]) in response to the viral infection [107]. In HBV, it has been shown that this activation stimulates KC to release chemokines and cytokines that attract other leukocytes [109]. Another feature of acute hepatitis pathology is ballooning degeneration, which is swelling of hepatocytes [107][110].
Distinct characteristics are likewise commonly seen in chronic hepatitis. Piecemeal/interface necrosis involves necrosis of periportal hepatocytes with accompanying inflammation involving the portal tract and periportal zone [111]. Necrosis can extend over multiple lobules, which is termed confluent necrosis. Conversely, focal necrosis involves a relatively small area of hepatocytes, with focal necrosis involving a smaller group of hepatocytes [111]. These histologic features, along with other findings common to chronic hepatitis including portal inflammation and fibrosis/cirrhosis, have been quantified [112]. The modified Histology Activity Index (HAI) [112] and METAVIR [113], which has been adapted for other forms of hepatitis besides hepatitis C virus [114], scoring systems are among the most widely used [115]. Of note, chronic HDV patients often suffer more severe histologic changes than those seen in chronic HBV patients with similar characteristics [116]. This is due to the fact that patients with chronic HDV are much more likely than chronic HBV patients to progress to more severe liver disease [65].
This entry is adapted from the peer-reviewed paper 10.3390/ijms232415973
References
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555.
- Zu Siederdissen, C.H.; Maasoumy, B.; Cornberg, M. What is new on HBsAg and other diagnostic markers in HBV infection? Best Pract. Res. Clin. Gastroenterol. 2017, 31, 281–289.
- World Health Organization. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b#:~:text=WHO%20estimates%20that%20296%20million,carcinoma%20(primary%20liver%20cancer) (accessed on 13 November 2022).
- MacLachlan, J.H.; Cowie, B.C. Hepatitis B virus epidemiology. Cold Spring Harb. Perspect. Med. 2015, 5, a021410.
- Shao, E.R.; Mboya, I.B.; Gunda, D.W.; Ruhangisa, F.G.; Temu, E.M.; Nkwama, M.L.; Pyuza, J.J.; Kilonzo, K.G.; Lyamuya, F.S.; Maro, V.P. Seroprevalence of hepatitis B virus infection and associated factors among healthcare workers in northern Tanzania. BMC Infect. Dis. 2018, 18, 474.
- Candotti, D.; Allain, J.P. Transfusion-transmitted hepatitis B virus infection. J. Hepatol. 2009, 51, 798–809.
- Liang, T.J. Hepatitis B: The virus and disease. Hepatology 2009, 49, S13–S21.
- Trepo, C.; Guillevin, L. Polyarteritis nodosa and extrahepatic manifestations of HBV infection: The case against autoimmune intervention in pathogenesis. J. Autoimmun. 2001, 16, 269–274.
- Liu, Y.; Shi, C.; Fan, J.; Wang, B.; Li, G. Hepatitis B-related glomerulonephritis and optimization of treatment. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 113–125.
- Mazzaro, C.; Dal Maso, L.; Visentini, M.; Ermacora, A.; Tonizzo, M.; Gattei, V.; Andreone, P. Recent news in the treatment of hepatitis B virus-related cryogobulinemic vasculitis. Minerva Med. 2020, 111, 566–572.
- Chen, Y.; Tian, Z. HBV-Induced Immune Imbalance in the Development of HCC. Front. Immunol. 2019, 10, 2048.
- Baig, S.; Alamgir, M. The extrahepatic manifestations of hepatitis B virus. J. Coll. Physicians Surg. Pak. 2008, 18, 451–457.
- Kim, G.W.; Imam, H.; Khan, M.; Mir, S.A.; Kim, S.J.; Yoon, S.K.; Hur, W.; Siddiqui, A. HBV-Induced Increased N6 Methyladenosine Modification of PTEN RNA Affects Innate Immunity and Contributes to HCC. Hepatology 2021, 73, 533–547.
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2022, 22, 19–32.
- Zhang, Y.Y.; Hu, K.Q. Rethinking the pathogenesis of hepatitis B virus (HBV) infection. J. Med. Virol. 2015, 87, 1989–1999.
- Engle, R.E.; De Battista, D.; Danoff, E.J.; Nguyen, H.; Chen, Z.; Lusso, P.; Purcell, R.H.; Farci, P. Distinct Cytokine Profiles Correlate with Disease Severity and Outcome in Longitudinal Studies of Acute Hepatitis B Virus and Hepatitis D Virus Infection in Chimpanzees. mBio 2020, 11, e02580-20.
- Paik, J.M.; Golabi, P.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Changes in the Global Burden of Chronic Liver Diseases from 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020, 72, 1605–1616.
- Pujol, F.; Jaspe, R.C.; Loureiro, C.L.; Chemin, I. Hepatitis B virus American genotypes: Pathogenic variants? Clin. Res. Hepatol. Gastroenterol. 2020, 44, 825–835.
- Lin, C.L.; Kao, J.H. Natural history of acute and chronic hepatitis B: The role of HBV genotypes and mutants. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 249–255.
- Tanwar, S.; Dusheiko, G. Is there any value to hepatitis B virus genotype analysis? Curr. Gastroenterol. Rep. 2012, 14, 37–46.
- Tsushima, K.; Tsuge, M.; Hiraga, N.; Uchida, T.; Murakami, E.; Makokha, G.N.; Kurihara, M.; Nomura, M.; Hiyama, Y.; Fujino, H.; et al. Comparison of intracellular responses between HBV genotype A and C infection in human hepatocyte chimeric mice. J. Gastroenterol. 2019, 54, 650–659.
- Ito, K.; Yotsuyanagi, H.; Yatsuhashi, H.; Karino, Y.; Takikawa, Y.; Saito, T.; Arase, Y.; Imazeki, F.; Kurosaki, M.; Umemura, T.; et al. Risk factors for long-term persistence of serum hepatitis B surface antigen following acute hepatitis B virus infection in Japanese adults. Hepatology 2014, 59, 89–97.
- Chan, H.L.; Hui, A.Y.; Wong, M.L.; Tse, A.M.; Hung, L.C.; Wong, V.W.; Sung, J.J. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 2004, 53, 1494–1498.
- Visvanathan, K.; Lang, T.; Ryan, K.; Wilson, R.; Skinner, N.A.; Thompson, A.J.; Ahn, S.H.; Weilert, F.; Abbott, W.; Gane, E.; et al. Toll-IL1 receptor-mediated innate immune responses vary across HBV genotype and predict treatment response to pegylated-IFN in HBeAg-positive CHB patients. J. Viral Hepat. 2016, 23, 170–179.
- Chiu, S.M.; Kuo, Y.H.; Wang, J.H.; Hung, C.H.; Hu, T.H.; Lu, S.N.; Chen, C.H. Associations of HBV Genotype B vs. C Infection with Relapse after Cessation of Entecavir or Tenofovir Therapy. Clin. Gastroenterol. Hepatol. 2020, 18, 2989–2997.e2983.
- Negro, F. Hepatitis D virus coinfection and superinfection. Cold Spring Harb. Perspect. Med. 2014, 4, a021550.
- Perez-Vargas, J.; Amirache, F.; Boson, B.; Mialon, C.; Freitas, N.; Sureau, C.; Fusil, F.; Cosset, F.L. Enveloped viruses distinct from HBV induce dissemination of hepatitis D virus in vivo. Nat. Commun. 2019, 10, 2098.
- Ferrante, N.D.; Lo Re, V., 3rd. Epidemiology, Natural History, and Treatment of Hepatitis Delta Virus Infection in HIV/Hepatitis B Virus Coinfection. Curr. HIV/AIDS Rep. 2020, 17, 405–414.
- Miao, Z.; Zhang, S.; Ou, X.; Li, S.; Ma, Z.; Wang, W.; Peppelenbosch, M.P.; Liu, J.; Pan, Q. Estimating the Global Prevalence, Disease Progression, and Clinical Outcome of Hepatitis Delta Virus Infection. J. Infect. Dis. 2020, 221, 1677–1687.
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.R.; Giorgi, E.; Kyomuhangi, I.; de Martel, C.; Hutin, Y.; Geretti, A.M. The global prevalence of hepatitis D virus infection: Systematic review and meta-analysis. J. Hepatol. 2020, 73, 523–532.
- Chen, H.Y.; Shen, D.T.; Ji, D.Z.; Han, P.C.; Zhang, W.M.; Ma, J.F.; Chen, W.S.; Goyal, H.; Pan, S.; Xu, H.G. Prevalence and burden of hepatitis D virus infection in the global population: A systematic review and meta-analysis. Gut 2019, 68, 512–521.
- Niro, G.A.; Ferro, A.; Cicerchia, F.; Brascugli, I.; Durazzo, M. Hepatitis delta virus: From infection to new therapeutic strategies. World J. Gastroenterol. 2021, 27, 3530–3542.
- Sellier, P.O.; Maylin, S.; Brichler, S.; Bercot, B.; Lopes, A.; Chopin, D.; Pogliaghi, M.; Munier, A.L.; Delcey, V.; Simoneau, G.; et al. Hepatitis B Virus-Hepatitis D Virus mother-to-child co-transmission: A retrospective study in a developed country. Liver Int. 2018, 38, 611–618.
- Zhang, Z.; Urban, S. Interplay between Hepatitis D Virus and the Interferon Response. Viruses 2020, 12, 1334.
- Roulot, D.; Brichler, S.; Layese, R.; BenAbdesselam, Z.; Zoulim, F.; Thibault, V.; Scholtes, C.; Roche, B.; Castelnau, C.; Poynard, T.; et al. Origin, HDV genotype and persistent viremia determine outcome and treatment response in patients with chronic hepatitis delta. J. Hepatol. 2020, 73, 1046–1062.
- Tseligka, E.D.; Clement, S.; Negro, F. HDV Pathogenesis: Unravelling Ariadne’s Thread. Viruses 2021, 13, 778.
- Mason, A.; Wick, M.; White, H.; Perrillo, R. Hepatitis B virus replication in diverse cell types during chronic hepatitis B virus infection. Hepatology 1993, 18, 781–789.
- Verrier, E.R.; Colpitts, C.C.; Schuster, C.; Zeisel, M.B.; Baumert, T.F. Cell Culture Models for the Investigation of Hepatitis B and D Virus Infection. Viruses 2016, 8, 261.
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 3, e00049.
- Iwamoto, M.; Watashi, K.; Tsukuda, S.; Aly, H.H.; Fukasawa, M.; Fujimoto, A.; Suzuki, R.; Aizaki, H.; Ito, T.; Koiwai, O.; et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun. 2014, 443, 808–813.
- Mason, W.S. Animal models and the molecular biology of hepadnavirus infection. Cold Spring Harb. Perspect. Med. 2015, 5, a021352.
- Giersch, K.; Dandri, M. In Vivo Models of HDV Infection: Is Humanizing NTCP Enough? Viruses 2021, 13, 588.
- Ohashi, K.; Marion, P.L.; Nakai, H.; Meuse, L.; Cullen, J.M.; Bordier, B.B.; Schwall, R.; Greenberg, H.B.; Glenn, J.S.; Kay, M.A. Sustained survival of human hepatocytes in mice: A model for in vivo infection with human hepatitis B and hepatitis delta viruses. Nat. Med. 2000, 6, 327–331.
- Du, Y.; Broering, R.; Li, X.; Zhang, X.; Liu, J.; Yang, D.; Lu, M. In Vivo Mouse Models for Hepatitis B Virus Infection and Their Application. Front. Immunol. 2021, 12, 766534.
- Bonamassa, B.; Hai, L.; Liu, D. Hydrodynamic gene delivery and its applications in pharmaceutical research. Pharm. Res. 2011, 28, 694–701.
- Chang, J.; Sigal, L.J.; Lerro, A.; Taylor, J. Replication of the human hepatitis delta virus genome Is initiated in mouse hepatocytes following intravenous injection of naked DNA or RNA sequences. J. Virol. 2001, 75, 3469–3473.
- Liu, Y.; Maya, S.; PLoSs, A. Animal Models of Hepatitis B Virus Infection-Success, Challenges, and Future Directions. Viruses 2021, 13, 777.
- Heermann, K.H.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W.H. Large surface proteins of hepatitis B virus containing the pre-s sequence. J. Virol. 1984, 52, 396–402.
- Selzer, L.; Zlotnick, A. Assembly and Release of Hepatitis B Virus. Cold Spring Harb. Perspect. Med. 2015, 5, a021394.
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antiviral Res. 2020, 182, 104925.
- Seeger, C.; Zoulim, F.; Masson, W.S. Hepadnaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2185–2221.
- Wei, L.; PLoss, A. Hepatitis B virus cccDNA is formed through distinct repair processes of each strand. Nat. Commun. 2021, 12, 1591.
- Been, M.D. HDV ribozymes. Curr. Top Microbiol. Immunol. 2006, 307, 47–65.
- Mentha, N.; Clement, S.; Negro, F.; Alfaiate, D. A review on hepatitis D: From virology to new therapies. J. Adv. Res. 2019, 17, 3–15.
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoude, G.; et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016, 63, 35–48.
- Perez-Vargas, J.; Teppa, E.; Amirache, F.; Boson, B.; de Oliveira, R.P.; Combet, C.; Bockmann, A.; Fusil, F.; Freitas, N.; Carbone, A.; et al. A fusion peptide in preS1 and the human protein disulfide isomerase ERp57 are involved in hepatitis B virus membrane fusion process. Elife 2021, 10, e64507.
- Herrscher, C.; Roingeard, P.; Blanchard, E. Hepatitis B Virus Entry into Cells. Cells 2020, 9, 1486.
- Wei, L.; PLoss, A. Core components of DNA lagging strand synthesis machinery are essential for hepatitis B virus cccDNA formation. Nat. Microbiol. 2020, 5, 715–726.
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984.
- Lin, N.; Ye, A.; Lin, J.; Liu, C.; Huang, J.; Fu, Y.; Wu, S.; Xu, S.; Wang, L.; Ou, Q. Diagnostic Value of Detection of Pregenomic RNA in Sera of Hepatitis B Virus-Infected Patients with Different Clinical Outcomes. J. Clin. Microbiol. 2020, 58, e01275-19.
- Revill, P.A.; Locarnini, S.A. New perspectives on the hepatitis B virus life cycle in the human liver. J. Clin. Investig. 2016, 126, 833–836.
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210.
- Tsai, K.N.; Kuo, C.F.; Ou, J.J. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol. 2018, 26, 33–42.
- Taylor, J.M.; Purcell, R.H.; Farci, P. Hepatitis D (Delta) Virus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2222–2241.
- Urban, S.; Neumann-Haefelin, C.; Lampertico, P. Hepatitis D virus in 2021: Virology, immunology and new treatment approaches for a difficult-to-treat disease. Gut 2021, 70, 1782–1794.
- Lo, K.; Hwang, S.B.; Duncan, R.; Trousdale, M.; Lai, M.M. Characterization of mRNA for hepatitis delta antigen: Exclusion of the full-length antigenomic RNA as an mRNA. Virology 1998, 250, 94–105.
- Hartwig, D.; Schutte, C.; Warnecke, J.; Dorn, I.; Hennig, H.; Kirchner, H.; Schlenke, P. The large form of ADAR 1 is responsible for enhanced hepatitis delta virus RNA editing in interferon-alpha-stimulated host cells. J. Viral Hepat. 2006, 13, 150–157.
- Dziri, S.; Rodriguez, C.; Gerber, A.; Brichler, S.; Alloui, C.; Roulot, D.; Deny, P.; Pawlotsky, J.M.; Gordien, E.; Le Gal, F. Variable In Vivo Hepatitis D Virus (HDV) RNA Editing Rates According to the HDV Genotype. Viruses 2021, 13, 1572.
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature 1986, 323, 508–514.
- Rizzetto, M. The adventure of delta. Liver Int. 2016, 36 (Suppl. S1), 135–140.
- Goodrum, G.; Pelchat, M. Insight into the Contribution and Disruption of Host Processes during HDV Replication. Viruses 2018, 11, 21.
- Filipovska, J.; Konarska, M.M. Specific HDV RNA-templated transcription by pol II in vitro. RNA 2000, 6, 41–54.
- Chang, J.; Nie, X.; Chang, H.E.; Han, Z.; Taylor, J. Transcription of hepatitis delta virus RNA by RNA polymerase II. J. Virol. 2008, 82, 1118–1127.
- Greco-Stewart, V.S.; Schissel, E.; Pelchat, M. The hepatitis delta virus RNA genome interacts with the human RNA polymerases I and III. Virology 2009, 386, 12–15.
- Li, Y.J.; Macnaughton, T.; Gao, L.; Lai, M.M. RNA-templated replication of hepatitis delta virus: Genomic and antigenomic RNAs associate with different nuclear bodies. J. Virol. 2006, 80, 6478–6486.
- Luo, G.X.; Chao, M.; Hsieh, S.Y.; Sureau, C.; Nishikura, K.; Taylor, J. A specific base transition occurs on replicating hepatitis delta virus RNA. J. Virol. 1990, 64, 1021–1027.
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123.
- Jayan, G.C.; Casey, J.L. Inhibition of hepatitis delta virus RNA editing by short inhibitory RNA-mediated knockdown of ADAR1 but not ADAR2 expression. J. Virol. 2002, 76, 12399–12404.
- Glenn, J.S.; Watson, J.A.; Havel, C.M.; White, J.M. Identification of a prenylation site in delta virus large antigen. Science 1992, 256, 1331–1333.
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on Prevention, Diagnosis, and Treatment of Chronic Hepatitis B: AASLD 2018 Hepatitis B Guidance. Clin. Liver Dis. 2018, 12, 33–34.
- McMahon, B.J. The natural history of chronic hepatitis B virus infection. Hepatology 2009, 49, S45–S55.
- Shapiro, C.N. Epidemiology of hepatitis B. Pediatr. Infect. Dis. J. 1993, 12, 433–437.
- van Santen, D.K.; Boyd, A.; Bruisten, S.; Sonder, G.J.; Prins, M.; van Houdt, R. Frequent delayed spontaneous seroclearance of hepatitis B virus after incident HBV infection among adult high-risk groups. J. Viral Hepat. 2020, 27, 81–87.
- Veronese, P.; Dodi, I.; Esposito, S.; Indolfi, G. Prevention of vertical transmission of hepatitis B virus infection. World J. Gastroenterol. 2021, 27, 4182–4193.
- Stevens, C.E.; Toy, P.T.; Tong, M.J.; Taylor, P.E.; Vyas, G.N.; Nair, P.V.; Gudavalli, M.; Krugman, S. Perinatal hepatitis B virus transmission in the United States. Prevention by passive-active immunization. JAMA 1985, 253, 1740–1745.
- Taylor, J.M. Infection by Hepatitis Delta Virus. Viruses 2020, 12, 648.
- Sagnelli, C.; Sagnelli, E.; Russo, A.; Pisaturo, M.; Occhiello, L.; Coppola, N. HBV/HDV Co-Infection: Epidemiological and Clinical Changes, Recent Knowledge and Future Challenges. Life 2021, 11, 169.
- Romeo, R. Hepatitis Delta: Natural history and outcome. Clin. Liver Dis. 2013, 2, 235–236.
- Botelho-Souza, L.F.; Vasconcelos, M.P.A.; Dos Santos, A.O.; Salcedo, J.M.V.; Vieira, D.S. Hepatitis delta: Virological and clinical aspects. Virol. J. 2017, 14, 177.
- Masood, U.; John, S. Hepatitis D. In StatPearls; NCBI Bookshelf: Bethesda, MD, USA, 2021.
- Hsieh, T.H.; Liu, C.J.; Chen, D.S.; Chen, P.J. Natural course and treatment of hepatitis D virus infection. J. Formos. Med. Assoc. 2006, 105, 869–881.
- Genesca, J.; Jardi, R.; Buti, M.; Vives, L.; Prat, S.; Esteban, J.I.; Esteban, R.; Guardia, J. Hepatitis B virus replication in acute hepatitis B, acute hepatitis B virus-hepatitis delta virus coinfection and acute hepatitis delta superinfection. Hepatology 1987, 7, 569–572.
- Giersch, K.; Dandri, M. Hepatitis B and Delta Virus: Advances on Studies about Interactions between the Two Viruses and the Infected Hepatocyte. J. Clin. Transl. Hepatol. 2015, 3, 220–229.
- Schaper, M.; Rodriguez-Frias, F.; Jardi, R.; Tabernero, D.; Homs, M.; Ruiz, G.; Quer, J.; Esteban, R.; Buti, M. Quantitative longitudinal evaluations of hepatitis delta virus RNA and hepatitis B virus DNA shows a dynamic, complex replicative profile in chronic hepatitis B and D. J. Hepatol. 2010, 52, 658–664.
- Braga, W.S.; de Oliveira, C.M.; de Araujo, J.R.; Mda, C.C.; Rocha, J.M.; Gimaque, J.B.; Silva, M.L.; Vasconcelos, H.L.; Ramasawmy, R.; Parana, R. Chronic HDV/HBV co-infection: Predictors of disease stage---A case series of HDV-3 patients. J. Hepatol. 2014, 61, 1205–1211.
- Mhlanga, A.; Zakh, R.; Churkin, A.; Reinharz, V.; Glenn, J.S.; Etzion, O.; Cotler, S.J.; Yurdaydin, C.; Barash, D.; Dahari, H. Modeling the Interplay between HDV and HBV in Chronic HDV/HBV Patients. Mathematics 2022, 10, 3917.
- Yurdaydin, C.; Keskin, O.; Kalkan, C.; Karakaya, F.; Caliskan, A.; Karatayli, E.; Karatayli, S.; Bozdayi, A.M.; Koh, C.; Heller, T.; et al. Optimizing lonafarnib treatment for the management of chronic delta hepatitis: The LOWR HDV-1 study. Hepatology 2018, 67, 1224–1236.
- Shekhtman, L.; Cotler, S.J.; Hershkovich, L.; Uprichard, S.L.; Bazinet, M.; Pantea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Krawczyk, A.; et al. Modelling hepatitis D virus RNA and HBsAg dynamics during nucleic acid polymer monotherapy suggest rapid turnover of HBsAg. Sci. Rep. 2020, 10, 7837.
- Guedj, J.; Rotman, Y.; Cotler, S.J.; Koh, C.; Schmid, P.; Albrecht, J.; Haynes-Williams, V.; Liang, T.J.; Hoofnagle, J.H.; Heller, T.; et al. Understanding early serum hepatitis D virus and hepatitis B surface antigen kinetics during pegylated interferon-alpha therapy via mathematical modeling. Hepatology 2014, 60, 1902–1910.
- Koh, C.; Canini, L.; Dahari, H.; Zhao, X.; Uprichard, S.L.; Haynes-Williams, V.; Winters, M.A.; Subramanya, G.; Cooper, S.L.; Pinto, P.; et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: A proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect. Dis. 2015, 15, 1167–1174.
- Castelnau, C.; Le Gal, F.; Ripault, M.P.; Gordien, E.; Martinot-Peignoux, M.; Boyer, N.; Pham, B.N.; Maylin, S.; Bedossa, P.; Deny, P.; et al. Efficacy of peginterferon alpha-2b in chronic hepatitis delta: Relevance of quantitative RT-PCR for follow-up. Hepatology 2006, 44, 728–735.
- Manesis, E.K.; Schina, M.; Le Gal, F.; Agelopoulou, O.; Papaioannou, C.; Kalligeros, C.; Arseniou, V.; Manolakopoulos, S.; Hadziyannis, E.S.; Gault, E.; et al. Quantitative analysis of hepatitis D virus RNA and hepatitis B surface antigen serum levels in chronic delta hepatitis improves treatment monitoring. Antivir. Ther. 2007, 12, 381–388.
- Zachou, K.; Yurdaydin, C.; Drebber, U.; Dalekos, G.N.; Erhardt, A.; Cakaloglu, Y.; Degertekin, H.; Gurel, S.; Zeuzem, S.; Bozkaya, H.; et al. Quantitative HBsAg and HDV-RNA levels in chronic delta hepatitis. Liver Int. 2010, 30, 430–437.
- Wedemeyer, H.; Yurdaydin, C.; Dalekos, G.N.; Erhardt, A.; Cakaloglu, Y.; Degertekin, H.; Gurel, S.; Zeuzem, S.; Zachou, K.; Bozkaya, H.; et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N. Engl. J. Med. 2011, 364, 322–331.
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498.
- Yurdaydin, C.; Bozkaya, H.; Onder, F.O.; Senturk, H.; Karaaslan, H.; Akdogan, M.; Cetinkaya, H.; Erden, E.; Erkan-Esin, O.; Yalcin, K.; et al. Treatment of chronic delta hepatitis with lamivudine vs. lamivudine + interferon vs. interferon. J. Viral Hepat. 2008, 15, 314–321.
- Lefkowitch, J.H. Acute Viral Hepatitis. In Scheuer’s Liver Biopsy Interpretation, 10th ed.; Lefkowitch, J.H., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 89–107.
- Dou, L.; Shi, X.; He, X.; Gao, Y. Macrophage Phenotype and Function in Liver Disorder. Front. Immunol. 2019, 10, 3112.
- Boltjes, A.; Movita, D.; Boonstra, A.; Woltman, A.M. The role of Kupffer cells in hepatitis B and hepatitis C virus infections. J. Hepatol. 2014, 61, 660–671.
- Mani, H.; Kleiner, D.E. Liver biopsy findings in chronic hepatitis B. Hepatology 2009, 49, S61–S71.
- Krishna, M. Patterns of necrosis in liver disease. Clin. Liver Dis. 2017, 10, 53–56.
- Ishak, K.; Baptista, A.; Bianchi, L.; Callea, F.; De Groote, J.; Gudat, F.; Denk, H.; Desmet, V.; Korb, G.; MacSween, R.N.; et al. Histological grading and staging of chronic hepatitis. J. Hepatol. 1995, 22, 696–699.
- Bedossa, P. Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. The French METAVIR Cooperative Study Group. Hepatology 1994, 20, 15–20.
- Axley, P.; Mudumbi, S.; Sarker, S.; Kuo, Y.F.; Singal, A.K. Patients with stage 3 compared to stage 4 liver fibrosis have lower frequency of and longer time to liver disease complications. PLoS ONE 2018, 13, e0197117.
- Mohamadnejad, M.; Tavangar, S.M.; Sotoudeh, M.; Kosari, F.; Khosravi, M.; Geramizadeh, B.; Montazeri, G.; Estakhri, A.; Mirnasseri, M.M.; Fazlollahi, A.; et al. Histopathological Study of Chronic Hepatitis B: A Comparative Study of Ishak and METAVIR Scoring Systems. Int. J. Organ Transplant. Med. 2010, 1, 171–176.
- Liao, B.; Zhang, F.; Lin, S.; He, H.; Liu, Y.; Zhang, J.; Xu, Y.; Yi, J.; Chen, Y.; Liu, H.; et al. Epidemiological, clinical and histological characteristics of HBV/HDV co-infection: A retrospective cross-sectional study in Guangdong, China. PLoS ONE 2014, 9, e115888.
This entry is offline, you can click here to edit this entry!