Neurodegenerative diseases are currently incurable. Numerous experimental data accumulated over the past fifty years have brought us closer to understanding the molecular and cell mechanisms responsible for their development. It is known that the basis of neurodegenerations are proteinopathies, disorders in the structure and function of various proteins that lead to their aggregation and toxic effects on cells. The most common neurodegenerative proteinopathies are amyloidosis (amyloid extracellular plaques in AD), tauopathy (various dementias), α-synucleopathy (Lowy bodies in PD), prionopathy, and TDP-43 proteinopathy (in amyotrophic lateral sclerosis (ALS)).
- neurodegeneration
- neurodegenerative diseases
- polyglutamine diseases
- proteinopathies
- Huntington’s disease
- huntingtin
1. Introduction
Neurodegenerative diseases are an extensive group of human pathologies of various genesis, among which both hereditary diseases and pathologies developed de novo are described. These diseases develop over decades, so they are more common in older people. In many countries of the world in recent decades, especially in countries with developed economies, in parallel with the increase in average life expectancy there has been an increase in the number of neurodegenerative diseases. They have become one of the main problems that worsen the quality of life in old age. Despite the difference in clinical manifestations, the development of neurodegenerative diseases may be based on similar molecular and/or cellular mechanisms. Considerable attention has been paid to the elucidation of these mechanisms in the last fifty years, and significant progress has been made in understanding the causes of their occurrence at the molecular, cellular and organic levels. However, this knowledge is still insufficient not only for the prevention, but also for the treatment of neurodegenerative diseases. In addition, if researchers do not talk about hereditary or genetically determined pathologies, there is also a problem of diagnosis and early detection of sporadically developing neurodegenerations.
The most common and actively researched neurodegenerative diseases are Alzheimer’s (AD) and Parkinson’s (PD) diseases. There is a wide range of less common pathologies, including hereditary causes.
At the moment, it is known that the basis of neurodegenerations are proteinopathies, disorders in the structure and function of various proteins that lead to their aggregation and toxic effects on cells. The most common neurodegenerative proteinopathies are amyloidosis (amyloid extracellular plaques in AD), tauopathy (various dementias), α-synucleopathy (Lowy bodies in PD), prionopathy, and TDP-43 proteinopathy (in amyotrophic lateral sclerosis (ALS)). Among hereditary neuropathologies, a group of polyglutamine diseases can be distinguished caused by the repetition of the cytosine-adenine-guanine (CAG) trinucleotide in the coding segment of certain genes, which leads to the appearance of mutant proteins. Huntington’s disease (HD) is one of the best studied polyglutamine diseases [1].
The greatest difficulty for the timely clinical diagnosis of neurodegenerative diseases is their sporadic forms. They are associated with a variety of genetic factors and environmental influences. The causes of their occurrence remain largely unknown. In general, the development of sporadic forms of neurodegenerative diseases leads to the same cellular pathologies as in hereditary forms, including the accumulation of toxic proteins. Modern scientific research is aimed at identifying specific biomarkers that would select potential patients in the early stages of the disease before the first symptoms appear. Determining the risk group is of great importance, because now methods have been developed to slow down the development of the disease, and the earlier such therapy begins, the better for the patient. For instance, in a number of publications devoted to this problem, an increased level of methylation of risk genes associated with the development of neurodegenerative diseases is discussed [2][3][4].
2. Proteins Responsible for Neurodegenerative Diseases, Their Association with Cell Organelles and Involvement in Cellular Functions
Most neurodegenerative diseases are proteinopathies that occur as a result of disorders in the structure and function of certain proteins. These defective proteins disorganize the normal function of intracellular organelles, which ultimately leads to toxic damage to nerve cells.
2.1. Amyloid-β
AD is one of the most common and severe neurodegenerative diseases. It is considered to be proven that AD is amyloidosis by nature; the disease is characterized by the formation of extracellular amyloid plaques in the nervous tissue. It is believed that their accumulation leads to the development of neurodegenerative disorders in patients, however (and researchers cannot fail to note this), these long-established ideas can be revised [5].
It has been shown that Aβ is formed by the cleavage of Aβ precursor protein (APP) by β- and γ-secretase. A number of β-amyloid peptides with a length of 39 to 43 amino acid residues are formed, the hydrophobic nature of which promotes self-aggregation and neurotoxicity [6][7]. APP is an expressed transmembrane protein (reviewed in [8][9]) that appears to perform important physiological functions in the synapse. It has been suggested that APP may function as a receptor involved in transsynaptic signaling [10][11], and may also participate in adhesion [12][13].
Many studies in the field of understanding the functions of Aβ appear almost daily. Nevertheless, researchers still have little idea of how to look for approaches to the qualitative therapy of neurodegenerative diseases associated with amyloid accumulation. Perhaps it is after the discovery of native APP functions that an understanding of the occurrence mechanisms, timely diagnosis and prevention of these diseases will appear.
2.2. Tau-Protein
The cause of some dementias is considered to be tauopathy. In taupathies, neurofibrillary tangles (NFT) are formed from the insoluble hyperphosphorylated tau protein. This disorder is observed in patients with AD, PD, frontal temporal dementia, Pick’s disease, and others [14].
The tau protein, originally defined as MAP (microtubule-associated protein), has over time been discovered to possess many alternative functions. It has been shown to bind to the p150 subunit of the dynactin complex, which provides the interaction of dynein with cargo [15]. Some studies provide evidence that tau protein can interact with actin and affect its polymerization, as well as its interaction with microtubules [16][17][18]. Tau protein can also interact with the plasma membrane [19][20][21][22], with some proteins involved in signal transmission [23][24], and has a binding site to DNA and RNA [25][26].
The wide range of tau protein interactions with other proteins and cellular structures presents ample opportunities for research activities that may lead to the discovery of new treatments for neurodegenerative processes.
2.3. α-Synuclein
α-Synucleinopathies are the cause of dementia and, in particular, PD. Due to the central hydrophobic region, α-synuclein has a high tendency for aggregation. It forms an intermediate ring structure called a protofibrill, which eventually transforms into insoluble polymers or fibrils [27]. These insoluble fibrils are the main component of Lewy bodies, the accumulation of which is observed in the neurons’ cytoplasm in dementia, including PD. Lewy bodies are found in both sporadic and hereditary forms of this disease. It has been shown that the aggregation of α-synuclein can be influenced by many factors, such as mutations, post-translational modifications of the protein and even the dopamine effects [28][29].
Despite many studies, the exact function of α-synuclein remains unclear. Monomeric α-synuclein plays significant roles in synaptic signal transduction and synaptic vesicle recirculation [30], including influencing glutamate release [31].
From all of the above, it follows that α-synuclein performs a number of important functions in the cell and affects various cellular organelles. However, researchers have yet to determine its main role in normal cellular metabolism.
2.4. Prions
Prions (PrP) are unique pathogenic proteins that have the property of self-replication when acquiring an alternative conformation. Prion diseases, or transmissible spongiform encephalopathies, includes Creutzfeld–Jacob disease, Gerstmann–Sträussler–Scheinker syndrome, kuru, fatal insomnia, and variable protease-sensitive prionopathy. All these diseases are associated with the conformational transformation of the normal cellular PrP into the abnormal PrPSc [32] through a post-translational process during which PrP acquires a high β-sheet content [33] with subsequent accumulation in the brain and nervous tissue [34].
The participation of PrPC in the development of prion diseases has been studied quite well, but its normal physiological role still remains the subject of scientific discussion. According to accumulated data, it can influence some early processes of cell differentiation [35][36][37]. PrPC plays a role in the development and maintenance of the nervous system as a whole [38][39][40]. Thus, PrPC can play an active role in many different cellular processes and can influence a wide range of physiological phenomena in the body. However, to date, its exact function and relationship with many intracellular structures remains unexplored.
2.5. TAR DNA-Binding Protein 43
TAR DNA-binding protein 43 (TDP-43) is a highly conserved heterogeneous nuclear ribonucleoprotein that controls the transcription, splicing and RNA stability of certain genes (reviewed in [41]). This protein is involved in various aspects of cell proliferation and apoptosis [42]. TDP-43 proteinopathy has been found in such forms of dementia as frontotemporal lobar degeneration and amyotrophic lateral sclerosis [43][44]. In these diseases, various forms of TDP-43 accumulate in the cytoplasm [44]. It remains unclear what causes neurotoxicity in this case, whether it is the aggregates themselves or the lack of regulatory factors for nucleic acids. This example shows that such combinations can be of great importance for the development of other neurodegenerative pathologies associated with the accumulation of Aβ, huntingtin, α-synuclein, etc.
2.6. Mutant Proteins in Polyglutamine Diseases
Currently, a number of neurodegenerative diseases have been described that have hereditary genetically determined causes, for example, polyglutamine diseases. This group of polyglutamine diseases, caused by the expansion of trinucleotide cytosine–adenine–guanine (CAG), repeats in the coding segment of certain genes. Such expansion leads to the appearance of mRNA with abnormally long repetitive CAG triplets and respective proteins with polyglutamine tracts in the cells.
HD is the most studied among the polyglutamine neurodegenerative diseases. Its development is associated with a mutation in the HTT gene and the consequence of this mutation and defective huntingtin (HTT) expression, is the death of neurons, mostly striatal neurons.
It has been shown that HTT is associated with numerous partner proteins, interacts with many intracellular structures and is involved in many cellular processes; therefore, HD is a very perspective model for the investigation.
3. Huntingtin–Protein Interactions as the Basis of Intracellular Pathologies and Potential Target for Therapeutic Intervention in Huntington Disease
HTT is a major cellular protein. As one of the research teams aptly said, huntingtin is ‘here, there, everywhere’ [45]. The HTT gene expresses in all tissues and organs in humans and mice, but HTT expression level is the highest in neural tissue [45]. Autosomal dominant mutation in the HTT causes an increase in the polyglutamine fragment length (encoded by the CAG codon) at the protein N-terminus and leads to the development of HD, a severe and currently incurable neurodegenerative pathology [1][46][47].
Despite intensive investigations, the functions of both mutant (mHTT) and wild-type HTT remain not fully understood. HTT can play an active role in cell physiology, being involved in a wide range of intracellular processes, such as cell transport, endocytosis, protein degradation and other cellular and molecular processes. HTT, as well as mHTT, interacts with different cell organelles directly or through their associated proteins (Figure 1). It is assumed that the effect of HTT on transport is due to its interaction via HAP1 (huntingtin-associated protein 1) with the p150Glued dinactin subunit and the kinesin family protein KIF5C [48][49]. HTT also interacts with both N- and C-terminal domains with dynamite 1 [45][50][51]. Researchers' own experimental data give reason to believe that a significant part of the HTT-involved cellular processes is mediated by microtubules and other cell cytoskeleton structures [52][53].
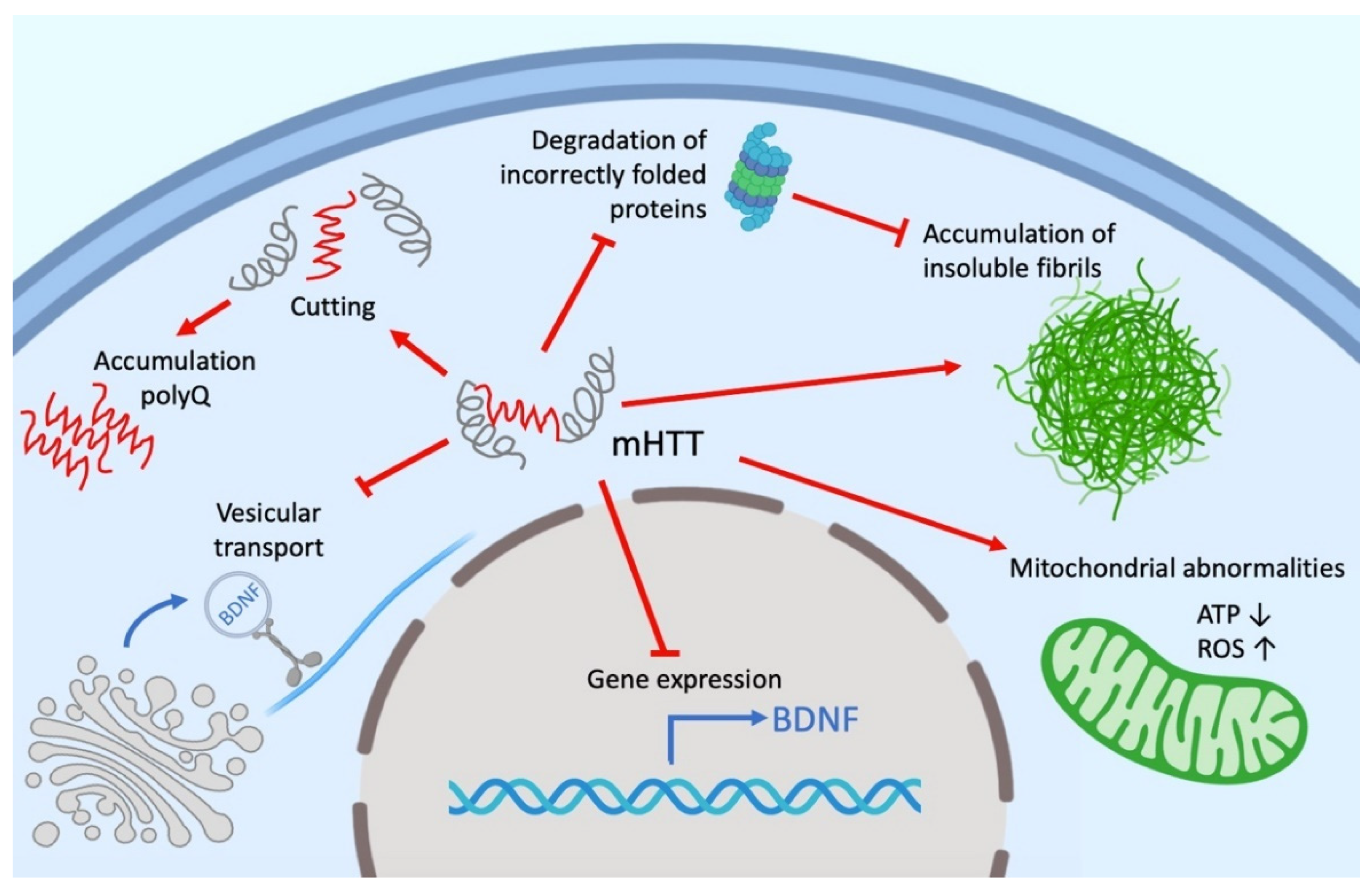
According to many studies, the number of HTT partner proteins can range from 100 to 350 [50][54][55]. The large number of molecular interactions, the nature of partner proteins, the relatively large size and stability of HTT suggest its role as a scaffold for a variety of protein complexes. This is also evidenced by the presence of sites in HTT such as HEAT domains. Thus, HTT function may be to coordinate cellular processes as a central component of several protein complexes [56].
4. The Spectrum of Possible Cellular Pathologies in Neurodegenerative Diseases
Experimental data indicate the involvement of many cellular processes in the neurodegenerative diseases development. These processes can be both unique to one disease and characteristic of many neurodegenerations. There are many discussions in the literature about phenomena such as the accumulation of protein aggregates or toxic protein fragments, violation of protein proteolysis, mRNA toxicity, mitochondrial disorders, nuclear violations, DNA damage, changes in protein expression and disruption of cellular transport.
The common manifestations of neurodegenerative diseases also include activation of microglia, cytokines, reactive astrogliosis and the launch of a broad inflammatory immune response [57][58]. These processes at the level of the organism lead to biochemical and structural changes in the surrounding neurons.
Summarizing numerous experimental data (but, of course, without claiming to cover them completely), researchers tried to determine which intracellular structures and cytophysiological processes mainly undergo pathological changes during the development of the most common neurodegenerative diseases (Figure 2).
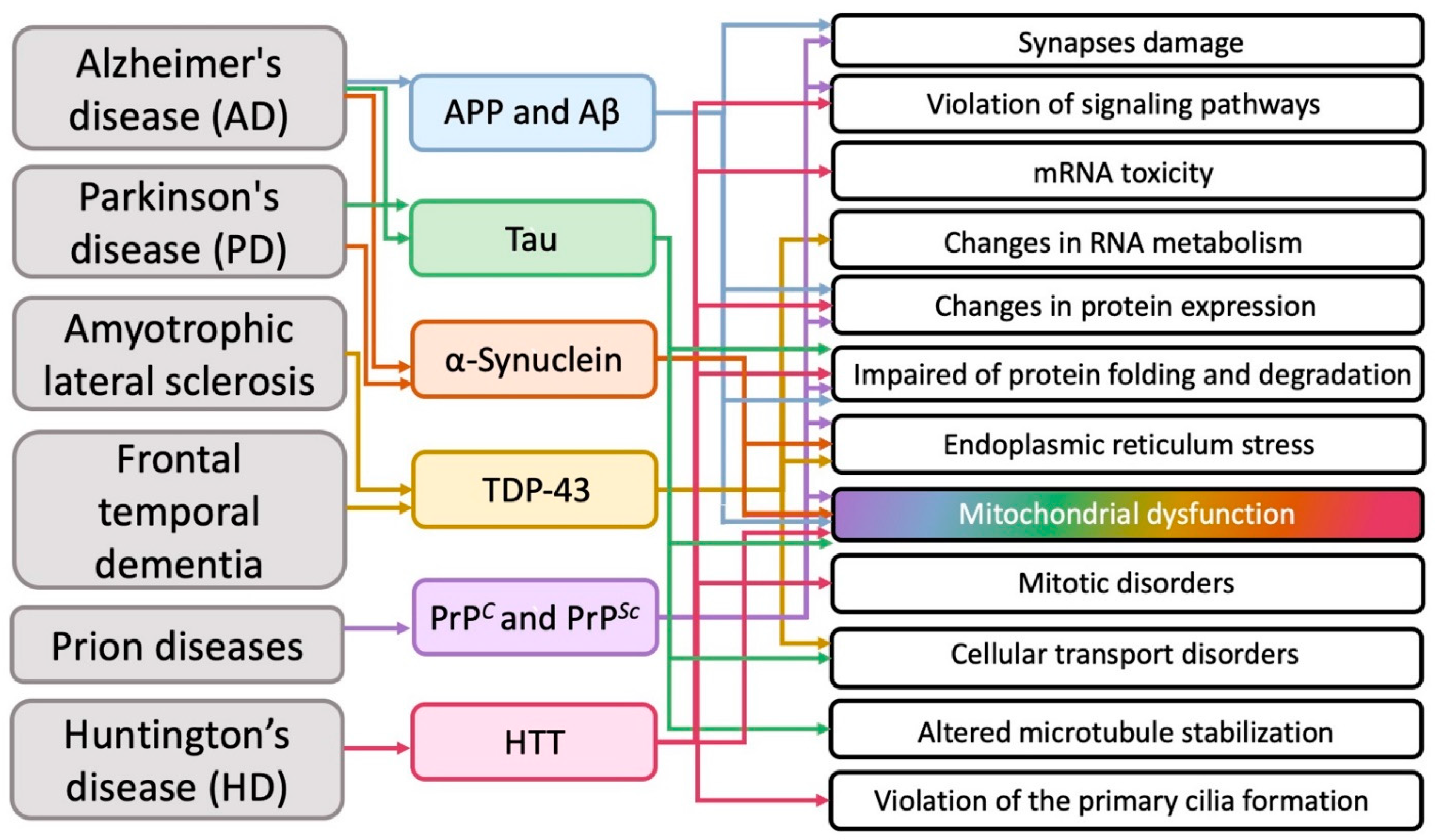
Figure 2. The main cellular pathologies developing during neurodegeneration.
It transpired that in various neurodegenerative diseases on the subcellular structural level, in addition to the obvious general problems with the corresponding proteins (impaired of protein expression, folding and degradation), mitochondria most often suffer from (Figure 2).
The biological aspects of the influence of proteins involved in neurodegeneration remains a subject of debate. It is known that in AD Aβ inhibits some components of the respiratory chain, thus causing mitochondrial depolarization, and also causes an increase in calcium levels, which leads to oxidative stress [59][60][61]. Disruption of the Tau protein leads to an abnormal distribution of mitochondria [62], a violation of the balance of their fusion and division [63]. In addition, oxidative stress is also observed [64]. Binding of α-synuclein directly to the mitochondrial membrane has been demonstrated [65]. This causes inhibition of the respiratory chain complex I [66] and stimulation of the calcium signal [67]. mHTT leads to a decrease in the activity of some components of the respiratory chain and a decrease in the membrane potential of mitochondria, as well as the buffer capacity of calcium [68][69].
5. Modern Approaches to the Development of Methods for Neurodegenerative Diseases Treatment
Modern medicine commonly uses exclusively symptomatic therapy, which is not able to affect the dynamics of the pathological process. Therapeutic approaches are designed to eliminate the manifestations of neurological disorders and somatic disorders that have arisen as a result of the disease development. Treatment methods include drugs that affect the neurotransmitter component (cholinesterase inhibitors and glutamate regulators in AD; catechol O-methyltransferase (COMT) inhibitors, MAO-B inhibitors, anticholinergics, L-Dopa, dopamine agonists and MAO-B inhibitors in PD; dopamine antagonists in HD); various channel blockers in ALS and ataxia (neuroprotectors); antipsychotics; antioxidants; and sedatives and muscle relaxants for the relief of choreic symptoms in HD. The therapy used is not specific to certain forms of neurodegenerative diseases and nor have a specific point of application to the regulation mechanisms of a certain protein, as such a violation of the conformation or structure causes the disease development.
As it follows from the analysis of modern research data (Figure 2), mitochondria and protein degradation systems in cells are most often affected in neurodegenerative diseases. To date, it is obvious that such disorders are most likely not the cause, but the earliest consequence of the development of the neurodegenerative process. Since mitochondrial disorders and protein degradation anomalies that occur in this case are characteristic of all neurodegenerations, mitochondria and cellular systems involved in the degradation process can serve as a target for the development of new therapies, specifically aimed at restoring their normal functioning and protecting certain cellular organelles.
Examples of such approaches include trials of mitochondrial-targeted antioxidants, such as SkQ1 and MitoQ, against neurodegenerative diseases (well described in review [70]). It is possible that the effectiveness of the use of mitochondrial antioxidants will be much higher if they are used in the early stages of the disease, before the development of pronounced damage to neurons. Therefore, the development of methods for the early diagnosis of neurodegenerative diseases is indeed an extremely urgent task.
In recent years, innovative immunological, genetic engineering and molecular biological approaches to the treatment of neurodegenerative diseases have been developed in the laboratory. One of the promising ways to combat the progressive development of neuron death is the vaccination of patients against neurodegeneration proteins (Aβ, α-synuclein etc.) [71], as well as the use of antibodies specific to them [72]. Methods aimed at editing mutant genes are being developed for the treatment of hereditary forms of neurodegenerative diseases. Researchers are looking for approaches to correcting the mHTT gene in HD using the CRISPR/Cas9 system [73]. At the same time, methods aimed at reducing the level of mutant proteins in the cell by affecting expression are being actively developed. These strategies have huge potential for treatment. RNA-targeted methods include the use of synthetic antisense oligonucleotides that bind to a specific sequence of ribonucleic acid, which can reduce the translation of mRNA into a disease-causing protein [74]. The use of the RNA interference mechanism shows satisfactory results in HD [75][76][77]. Thus, the specific effect on neurodegeneration-associated proteins in this case is the key to the development of effective therapies. Finally, an approach based on the use of neurodegeneration suppressor proteins, which can be considered as potential targets for therapeutic intervention, has already been mentioned in this review [47] and seems very promising.
6. Conclusions
Obviously, it is impossible to offer a universal approach to the treatment of all neurodegenerative diseases. It is necessary to search for individual approaches to protect the structure and functions of specific proteins involved in the development of neurodegeneration at the cellular level. In this regard, promising are the studies conducted on the HD model, allowed to detect HTT-interacting proteins that are genetic modifiers in neurodegenerative disorders [50]. Non-functioning neurodegeneration suppressors found among modifiers can be considered potential targets for therapeutic intervention. The search for similar neurodegeneration suppressor proteins, among partner proteins interacting with mutant proteins involved in the development of other neurodegenerative processes, seems to be a promising direction for further research aimed at restoring the normal function of damaged proteins.
Thus, a combination of therapy aimed at maintaining mitochondrial function and any of the above innovative approaches aimed at editing mutant genes, at suppressing (by means of neurodegeneration suppressor proteins), or reducing the level of mutant proteins in the cell (for diseases that are not the result of a genetic mutation), currently seems to be the most promising approach for the treatment of these disorders. In researchers' opinion, taking into account the current state of research, the greatest progress regarding these approaches should be expected in areas related to the development of methods aimed at reducing/suppressing the level of mutant/defective proteins in the cell. They can provide a qualitative breakthrough in the treatment of neurodegenerative diseases.
This entry is adapted from the peer-reviewed paper 10.3390/ijms232415533
References
- MacMillan, J.C.; Snell, R.G.; Tyler, A.; Houlihan, G.D.; Fenton, I.; Cheadle, J.P.; Lazarou, L.P.; Shaw, J.D.; Harper, P.S. Molecular Analysis and Clinical Correlations of the Huntington’s Disease Mutation. Lancet 1993, 342, 954–958.
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-Adenosylmethionine/Homocysteine Cycle Alterations Modify DNA Methylation Status with Consequent Deregulation of PS1 and BACE and Beta-Amyloid Production. Mol. Cell. Neurosci. 2005, 28, 195–204.
- Wang, Q.; Chen, Y.; Readhead, B.; Chen, K.; Su, Y.; Reiman, E.M.; Dudley, J.T. Longitudinal Data in Peripheral Blood Confirm That PM20D1 Is a Quantitative Trait Locus (QTL) for Alzheimer’s Disease and Implicate Its Dynamic Role in Disease Progression. Clin. Epigenetics 2020, 12, 189.
- Pellegrini, C.; Pirazzini, C.; Sala, C.; Sambati, L.; Yusipov, I.; Kalyakulina, A.; Ravaioli, F.; Kwiatkowska, K.M.; Durso, D.F.; Ivanchenko, M.; et al. A Meta-Analysis of Brain DNA Methylation Across Sex, Age, and Alzheimer’s Disease Points for Accelerated Epigenetic Aging in Neurodegeneration. Front. Aging Neurosci. 2021, 13, 639428.
- Piller, C. Blots on a Field? Science 2022, 377, 358–363.
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890.
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249.
- Muller, U.C.; Zheng, H. Physiological Functions of APP Family Proteins. Cold Spring Harb. Perspect. Med. 2012, 2, a006288.
- Baumkötter, F.; Wagner, K.; Eggert, S.; Wild, K.; Kins, S. Structural Aspects and Physiological Consequences of APP/APLP Trans-Dimerization. Exp. Brain Res. 2012, 217, 389–395.
- Ho, A.; Südhof, T.C. Binding of F-Spondin to Amyloid-β Precursor Protein: A Candidate Amyloid-β Precursor Protein Ligand That Modulates Amyloid-β Precursor Protein Cleavage. Proc. Natl. Acad. Sci. USA 2004, 101, 2548–2553.
- Park, J.H.; Gimbel, D.A.; GrandPre, T.; Lee, J.-K.; Kim, J.-E.; Li, W.; Lee, D.H.S.; Strittmatter, S.M. Alzheimer Precursor Protein Interaction with the Nogo-66 Receptor Reduces Amyloid-β Plaque Deposition. J. Neurosci. 2006, 26, 1386–1395.
- Yamazaki, T.; Koo, E.H.; Selkoe, D.J. Cell Surface Amyloid β-Protein Precursor Colocalizes with Β1 Integrins at Substrate Contact Sites in Neural Cells. J. Neurosci. 1997, 17, 1004–1010.
- Young-Pearse, T.L.; Chen, A.C.; Chang, R.; Marquez, C.; Selkoe, D.J. Secreted APP Regulates the Function of Full-Length APP in Neurite Outgrowth through Interaction with Integrin Beta1. Neural Dev. 2008, 3, 15.
- Kovacs, G.G. Invited Review: Neuropathology of Tauopathies: Principles and Practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23.
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of Tau Protein with the Dynactin Complex. EMBO J. 2007, 26, 4546–4554.
- Selden, S.C.; Pollard, T.D. Phosphorylation of Microtubule-Associated Proteins Regulates Their Interaction with Actin Filaments. J. Biol. Chem. 1983, 258, 7064–7071.
- Yamauchi, P.S.; Purich, D.L. Microtubule-Associated Protein Interactions with Actin Filaments: Evidence for Differential Behavior of Neuronal MAP-2 and Tau in the Presence of Phosphatidylinositol. Biochem. Biophys. Res. Commun. 1993, 190, 710–715.
- Elie, A.; Prezel, E.; Guérin, C.; Denarier, E.; Ramirez-Rios, S.; Serre, L.; Andrieux, A.; Fourest-Lieuvin, A.; Blanchoin, L.; Arnal, I. Tau Co-Organizes Dynamic Microtubule and Actin Networks. Sci. Rep. 2015, 5, 9964.
- Brandt, R.; Léger, J.; Lee, G. Interaction of Tau with the Neural Plasma Membrane Mediated by Tau’s Amino-Terminal Projection Domain. J. Cell Biol. 1995, 131, 1327–1340.
- Arrasate, M.; Pérez, M.; Avila, J. Tau Dephosphorylation at Tau-1 Site Correlates with Its Association to Cell Membrane. Neurochem. Res. 2000, 25, 43–50.
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation Regulates Tau Interactions with Src Homology 3 Domains of Phosphatidylinositol 3-Kinase, Phospholipase Cγ1, Grb2, and Src Family Kinases. J. Biol. Chem. 2008, 283, 18177–18186.
- Gauthier-Kemper, A.; Weissmann, C.; Golovyashkina, N.; Sebö-Lemke, Z.; Drewes, G.; Gerke, V.; Heinisch, J.J.; Brandt, R. The Frontotemporal Dementia Mutation R406W Blocks Tau’s Interaction with the Membrane in an Annexin A2–Dependent Manner. J. Cell Biol. 2011, 192, 647–661.
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau Interacts with Src-Family Non-Receptor Tyrosine Kinases. J. Cell Sci. 1998, 111, 3167–3177.
- Usardi, A.; Pooler, A.M.; Seereeram, A.; Reynolds, C.H.; Derkinderen, P.; Anderton, B.; Hanger, D.P.; Noble, W.; Williamson, R. Tyrosine Phosphorylation of Tau Regulates Its Interactions with Fyn SH2 Domains, but Not SH3 Domains, Altering the Cellular Localization of Tau. FEBS J. 2011, 278, 2927–2937.
- Qi, H.; Cantrelle, F.-X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buée, L.; Lippens, G.; Bonnefoy, E.; Galas, M.-C.; Landrieu, I. Nuclear Magnetic Resonance Spectroscopy Characterization of Interaction of Tau with DNA and Its Regulation by Phosphorylation. Biochemistry 2015, 54, 1525–1533.
- Wang, X.S.; Wang, D.L.; Zhao, J.; Qu, M.H.; Zhou, X.H.; He, H.J.; He, R.Q. The Proline-Rich Domain and the Microtubule Binding Domain of Protein Tau Acting as RNA Binding Domains. Protein Pept. Lett. 2006, 13, 679–685.
- Giasson, B.I.; Murray, I.V.J.; Trojanowski, J.Q.; Lee, V.M.-Y. A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of α-Synuclein Is Essential for Filament Assembly. J. Biol. Chem. 2001, 276, 2380–2386.
- Conway, K.A.; Lee, S.-J.; Rochet, J.-C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of Oligomerization, Not Fibrillization, Is a Shared Property of Both α-Synuclein Mutations Linked to Early-Onset Parkinson’s Disease: Implications for Pathogenesis and Therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576.
- Conway, K.A.; Rochet, J.-C.; Bieganski, R.M.; Lansbury, P.T. Kinetic Stabilization of the α-Synuclein Protofibril by a Dopamine-α-Synuclein Adduct. Science 2001, 294, 1346–1349.
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins Regulate the Kinetics of Synaptic Vesicle Endocytosis. J. Neurosci. 2014, 34, 9364–9376.
- Gureviciene, I.; Gurevicius, K.; Tanila, H. Role of α-Synuclein in Synaptic Glutamate Release. Neurobiol. Dis. 2007, 28, 83–89.
- Prusiner, S.B. Novel Proteinaceous Infectious Particles Cause Scrapie. Science 1982, 216, 136–144.
- Safar, J.; Roller, P.P.; Gajdusek, D.C.; Gibbs, C.J. Conformational Transitions, Dissociation, and Unfolding of Scrapie Amyloid (Prion) Protein. J. Biol. Chem. 1993, 268, 20276–20284.
- Merz, P.A.; Somerville, R.A.; Wisniewski, H.M.; Iqbal, K. Abnormal Fibrils from Scrapie-Infected Brain. Acta Neuropathol. 1981, 54, 63–74.
- Lee, Y.J.; Baskakov, I.V. The Cellular Form of the Prion Protein Is Involved in Controlling Cell Cycle Dynamics, Self-Renewal, and the Fate of Human Embryonic Stem Cell Differentiation. J. Neurochem. 2013, 124, 310–322.
- Prodromidou, K.; Papastefanaki, F.; Sklaviadis, T.; Matsas, R. Functional Cross-Talk Between the Cellular Prion Protein and the Neural Cell Adhesion Molecule Is Critical for Neuronal Differentiation of Neural Stem/Precursor Cells. Stem Cells 2014, 32, 1674–1687.
- Halliez, S.; Martin-Lannerée, S.; Passet, B.; Hernandez-Rapp, J.; Castille, J.; Urien, C.; Chat, S.; Laude, H.; Vilotte, J.-L.; Mouillet-Richard, S.; et al. Prion Protein Localizes at the Ciliary Base during Neural and Cardiovascular Development and Its Depletion Affects α-Tubulin Post-Translational Modifications. Sci. Rep. 2015, 5, 17146.
- Adle-Biassette, H.; Verney, C.; Peoc’h, K.; Dauge, M.-C.; Razavi, F.; Choudat, L.; Gressens, P.; Budka, H.; Henin, D. Immunohistochemical Expression of Prion Protein (PrPC) in the Human Forebrain During Development. J. Neuropathol. Exp. Neurol. 2006, 65, 698–706.
- Lima, F.R.S.; Arantes, C.P.; Muras, A.G.; Nomizo, R.; Brentani, R.R.; Martins, V.R. Cellular Prion Protein Expression in Astrocytes Modulates Neuronal Survival and Differentiation. J. Neurochem. 2007, 103, 2164–2176.
- Skedsmo, F.S.; Malachin, G.; Våge, D.I.; Hammervold, M.M.; Salvesen, Ø.; Ersdal, C.; Ranheim, B.; Stafsnes, M.H.; Bartosova, Z.; Bruheim, P.; et al. Demyelinating Polyneuropathy in Goats Lacking Prion Protein. FASEB J. 2020, 34, 2359–2375.
- Buratti, E. Multiple Roles of TDP-43 in Gene Expression, Splicing Regulation, and Human Disease. Front. Biosci. 2008, 13, 867.
- Ayala, Y.M.; Misteli, T.; Baralle, F.E. TDP-43 Regulates Retinoblastoma Protein Phosphorylation through the Repression of Cyclin-Dependent Kinase 6 Expression. Proc. Natl. Acad. Sci. USA 2008, 105, 3785–3789.
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611.
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133.
- Sousa, C.M.; Humbert, S. Huntingtin: Here, There, Everywhere! J. Huntingt. Dis. 2013, 2, 395–403.
- Gusella, J.F.; Wexler, N.S.; Conneally, P.M.; Naylor, S.L.; Anderson, M.A.; Tanzi, R.E.; Watkins, P.C.; Ottina, K.; Wallace, M.R.; Sakaguchi, A.Y.; et al. A Polymorphic DNA Marker Genetically Linked to Huntington’s Disease. Nature 1983, 307, 234–238.
- Myers, R.H. Huntington’s Disease Genetics. NeuroRX 2004, 1, 255–262.
- Engelender, S.; Sharp, A.H.; Colomer, V.; Tokito, M.K.; Lanahan, A.; Worley, P.; Holzbaur, E.L.F.; Ross, C.A. Huntingtin-Associated Protein 1 (HAP1) Interacts with the P150Glued Bubunit of Dynactin. Hum. Mol. 1997, 6, 2205–2212.
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L.F. Huntingtin Facilitates Dynein/Dynactin-Mediated Vesicle Transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050.
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin Interacting Proteins Are Genetic Modifiers of Neurodegeneration. PLoS Genet. 2007, 3, 689–708.
- El-Daher, M.-T.; Hangen, E.; Bruyere, J.; Poizat, G.; Al-Ramahi, I.; Pardo, R.; Bourg, N.; Souquere, S.; Mayet, C.; Pierron, G.; et al. Huntingtin Proteolysis Releases Non-PolyQ Fragments That Cause Toxicity through Dynamin 1 Dysregulation. EMBO J. 2015, 34, 2255–2271.
- Taran, A.S.; Shuvalova, L.D.; Lagarkova, M.A.; Alieva, I.B. Huntington’s Disease-An Outlook on the Interplay of the HTT Protein, Microtubules and Actin Cytoskeletal Components. Cells 2020, 9, 1514.
- Taran, A.; Belikova, L.; Lavrushkina, S.; Bogomazova, A.; Lagarkova, M.; Alieva, I. Microtubule Plus-End Dynamics Visualization in Huntington’s Disease Model Based on Human Primary Skin Fibroblasts. J. Vis. Exp. 2022, 2022, e62963.
- Culver, B.P.; Savas, J.N.; Park, S.K.; Choi, J.H.; Zheng, S.; Zeitlin, S.O.; Yates, J.R.; Tanese, N. Proteomic Analysis of Wild-Type and Mutant Huntingtin-Associated Proteins in Mouse Brains Identifies Unique Interactions and Involvement in Protein Synthesis. J. Biol. Chem. 2012, 287, 21599–21614.
- Tourette, C.; Li, B.; Bell, R.; O’Hare, S.; Kaltenbach, L.S.; Mooney, S.D.; Hughes, R.E. A Large Scale Huntingtin Protein Interaction Network Implicates RHO GTPase Signaling Pathways in Huntington Disease. J. Biol. Chem. 2014, 289, 6709–6726.
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926.
- Myers, R.H.; Vonsattel, J.P.; Paskevich, P.A.; Kiely, D.K.; Stevens, T.J.; Cupples, L.A.; Richardson, E.P.; Bird, E.D. Decreased Neuronal and Increased Oligodendroglial Densities in Huntington’s Disease Caudate Nucleus. J. Neuropathol. Exp. Neurol. 1991, 50, 729–742.
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The Role of Glial Reaction and Inflammation in Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2006, 991, 214–228.
- Parks, J.K.; Smith, T.S.; Trimmer, P.A.; Bennett, J.P., Jr.; Parker, W.D., Jr. Neurotoxic Aβ Peptides Increase Oxidative Stress in Vivo through NMDA-Receptor and Nitric-Oxide-Synthase Mechanisms, and Inhibit Complex IV Activity and Induce a Mitochondrial Permeability Transition in Vitro. J. Neurochem. 2001, 76, 1050–1056.
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in Intracellular Calcium and Glutathione in Astrocytes as the Primary Mechanism of Amyloid Neurotoxicity. J. Neurosci. 2003, 23, 5088.
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. β-Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. J. Neurosci. 2004, 24, 565.
- Praprotnik, D.; Smith, M.A.; Richey, P.L.; Vinters, H.V.; Perry, G. Filament Heterogeneity within the Dystrophic Neurites of Senile Plaques Suggests Blockage of Fast Axonal Transport in Alzheimer’s Disease. Acta Neuropathol. 1996, 91, 226–235.
- Wang, X.; Su, B.; Lee, H.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer’s Disease. J. Neurosci. 2009, 29, 9090.
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial Hyperpolarization in IPSC-Derived Neurons from Patients of FTDP-17 with 10+16 MAPT Mutation Leads to Oxidative Stress and Neurodegeneration. Redox Biol. 2017, 12, 410–422.
- Nakamura, K.; Nemani, V.M.; Wallender, E.K.; Kaehlcke, K.; Ott, M.; Edwards, R.H. Optical Reporters for the Conformation of α-Synuclein Reveal a Specific Interaction with Mitochondria. J. Neurosci. 2008, 28, 12305.
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, Prefibrillar α-Synuclein Oligomers Promote Complex I-Dependent, Ca2+-Induced Mitochondrial Dysfunction. J. Biol. Chem. 2014, 289, 21490–21507.
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ Is a Key Factor in α-Synuclein-Induced Neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801.
- Benchoua, A.; Trioulier, Y.; Zala, D.; Gaillard, M.-C.; Lefort, N.; Dufour, N.; Saudou, F.; Elalouf, J.-M.; Hirsch, E.; Hantraye, P.; et al. Involvement of Mitochondrial Complex II Defects in Neuronal Death Produced by N-Terminus Fragment of Mutated Huntingtin. Mol. Biol. Cell 2006, 17, 1652–1663.
- Lim, D.; Fedrizzi, L.; Tartari, M.; Zuccato, C.; Cattaneo, E.; Brini, M.; Carafoli, E. Calcium Homeostasis and Mitochondrial Dysfunction in Striatal Neurons of Huntington Disease. J. Biol. Chem. 2008, 283, 5780–5789.
- Shinn, L.J.; Lagalwar, S. Treating Neurodegenerative Disease with Antioxidants: Efficacy of the Bioactive Phenol Resveratrol and Mitochondrial-Targeted MitoQ and SkQ. Antioxidants 2021, 10, 573.
- Song, G.; Yang, H.; Shen, N.; Pham, P.; Brown, B.; Lin, X.; Hong, Y.; Sinu, P.; Cai, J.; Li, X.; et al. An Immunomodulatory Therapeutic Vaccine Targeting Oligomeric Amyloid-β1. J. Alzheimer’s Dis. 2020, 77, 1639–1653.
- Walsh, S.; Merrick, R.; Milne, R.; Brayne, C. Aducanumab for Alzheimer’s Disease? BMJ 2021, 374, n1682.
- Shin, J.W.; Kim, K.-H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.-M. Permanent Inactivation of Huntington’s Disease Mutation by Personalized Allele-Specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576.
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316.
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L. RNA Interference Improves Motor and Neuropathological Abnormalities in a Huntington’s Disease Mouse Model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825.
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G.; et al. Therapeutic Silencing of Mutant Huntingtin with SiRNA Attenuates Striatal and Cortical Neuropathology and Behavioral Deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209.
- Yu, D.; Pendergraff, H.; Liu, J.; Kordasiewicz, H.B.; Cleveland, D.W.; Swayze, E.E.; Lima, W.F.; Crooke, S.T.; Prakash, T.P.; Corey, D.R. Single-Stranded RNAs Use RNAi to Potently and Allele-Selectively Inhibit Mutant Huntingtin Expression. Cell 2012, 150, 895–908.