Rare diseases, especially monogenic diseases, which usually affect a single target protein, have attracted growing interest in drug research by encouraging pharmaceutical companies to design and develop therapeutic products to be tested in the clinical arena. Acute intermittent porphyria (AIP) is one of these rare diseases. AIP is characterized by haploinsufficiency in the third enzyme of the heme biosynthesis pathway. Identification of the liver as the target organ and a detailed molecular characterization have enabled the development and approval of several therapies to manage this disease.
- rare metabolic diseases
- hemoproteins
- liver function
- mitochondrial cytochromes
1. Introduction
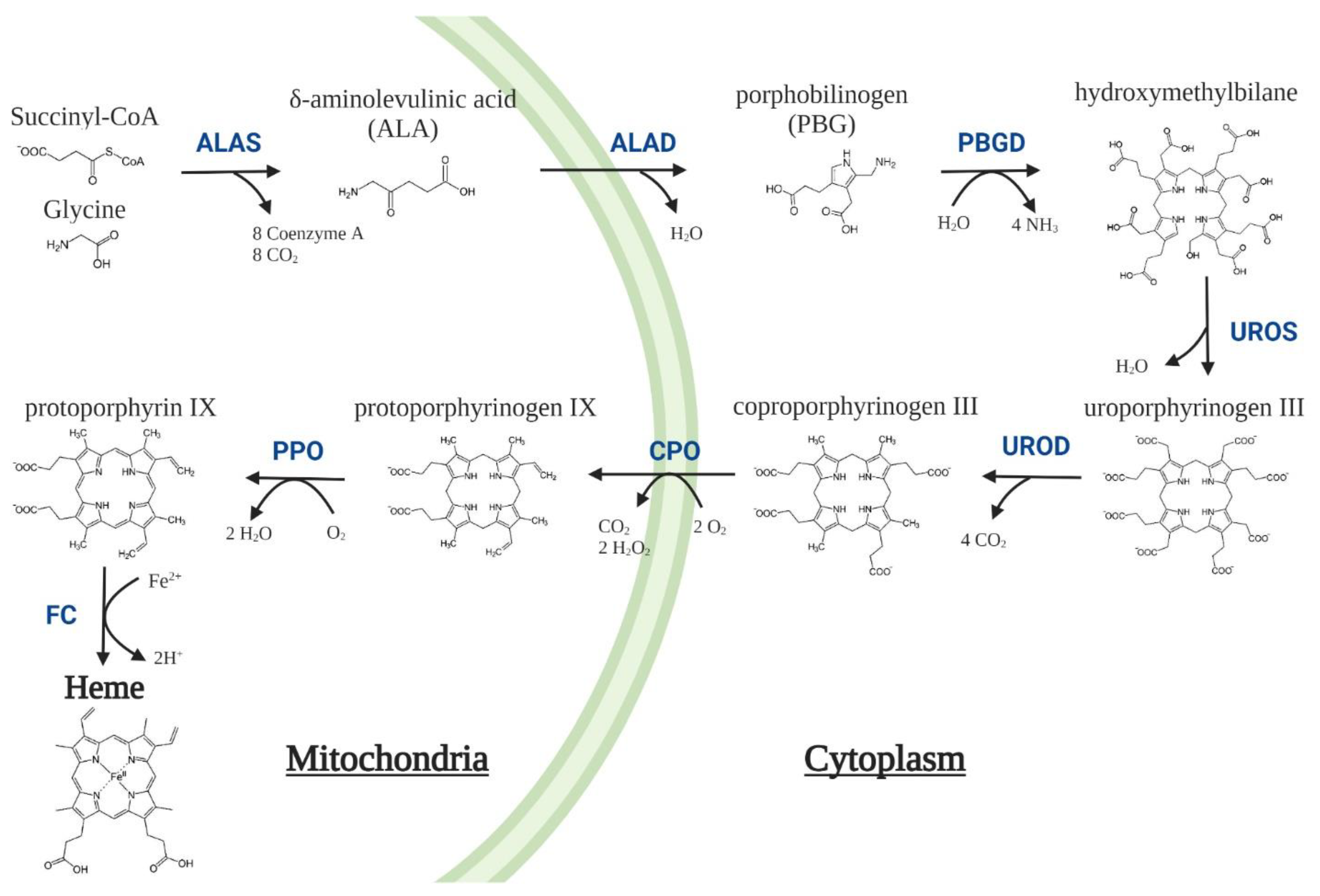
| Enzyme | Mutation | Disease | OMIM |
|---|---|---|---|
| δ-Aminolevulinic acid synthase 2 (ALAS2, EC 2.3.1.37) | GoF | X-linked Protoporphyria (XLP) | 300752 |
| δ-Aminolevulinic acid dehydratase (ALAD, EC 4.2.1.24) | LoF | ALAD Deficiency Porphyria (ADP) | 612740 |
| Porphobilinogen deaminase (PBGD, EC 2.5.1.61) | LoF | Acute Intermittent Porphyria (AIP) | 176000 |
| Uroporphyrinogen III synthase (UROS, EC 4.2.1.75) | LoF | Congenital Erythropoietic Porphyria (CEP) | 263700 |
| Uroporphyrinogen III decarboxylase (UROD, EC 4.1.1.37) | LoF | Porphyria Cutanea Tarda (PCT) Hepatoerythropoietic porphyria (HEP) |
176100 |
| Coproporphyrinogen oxidase(CPO, EC 1.3.3.3) | LoF | Hereditary Coproporphyria (HCP) | 121300 |
| Protoporphyrinogen oxidase(PPO, EC 1.3.3.4) | LoF | Variegate Porphyria (VP) | 176200 |
| Ferrochelatase(FC, EC 4.99.1.1) | LoF | Erythropoietic Protoporphyria (EPP) | 177000 |
2. Acute Intermittent Porphyria
Acute Neurovisceral Attacks
3. Current Treatments
- 2.
-
Patients suffering sporadic acute attacks (1 to 3 per year). Symptoms are very heterogeneous and include autonomous (intense pain typically in the abdomen that can also affect the back, legs, arms, or chest; nausea; vomiting; diarrhea/constipation; hypertension; and/or tachycardia), central (seizures, anxiety, depression, reduced consciousness, psychosis, insomnia, hallucinations or posterior reversible encephalopathy syndrome (PRES) on MRI scan, among others) and peripheral (muscle weakness, paralysis, reduced tendon reflexes) nervous system involvement. Severe neurological complications may cause death due to respiratory and bulbar paralysis[19].Carbohydrate overload (300 to 500 g/day, based on oral or iv glucose infusions) is recommended for the treatment of patients mild pain and no paresis. A mild attack can quickly become a severe attack, characterized by severe neuropathic abdominal and muscle pain, significant hyponatremia, urinary retention or incontinence, peripheral neuropathy (85% of sporadic AIP), or central nervous system (CNS) involvement. The treatment recommended for severe porphyria attacks consists of daily intravenous administration of hemin (hemin arginate, Normosang® in Europe and lyophilized hematin, Panhematin® in the US, both from Recordati, Milan, Italy) for a period of 4 days (3–4 mg/kg of hemin/day). It is more effective than glucose in reducing the formation of porphyrin precursors but is more expensive and is not available in all countries. Hemin therapy acts through retroinhibition of the ALAS1, which reduces production and accumulation of PBG and ALA (Figure 2). Hemin treatment lasted from one to four days, and biochemical remission of ALA and PBG is typically not produced until two or three days after the beginning of treatment. The reduction in abdominal pain is typically observed on the third day of treatment[20].
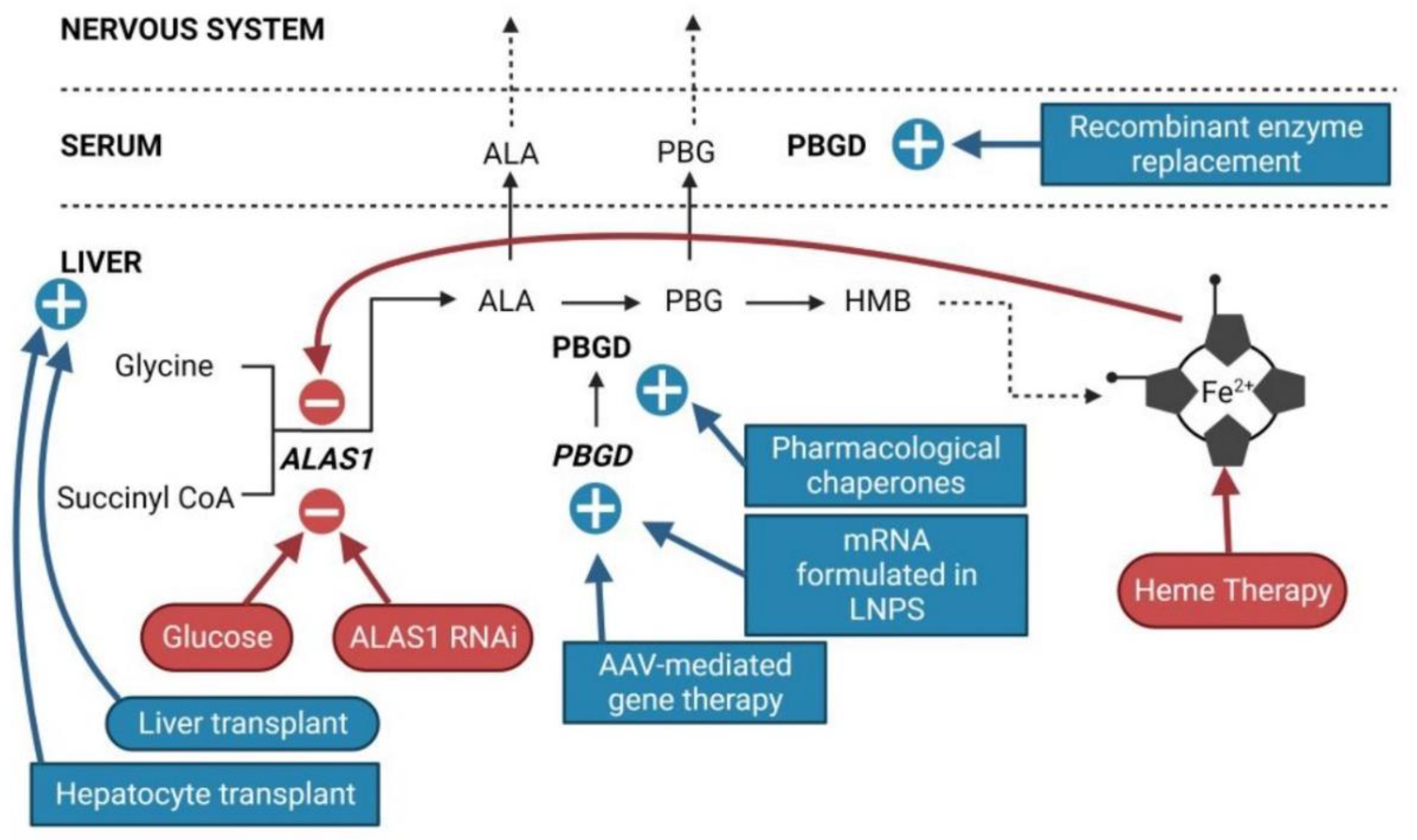
4. Innovative Therapies
The current prevalent R&D trends related to AIP therapy focus on increasing hepatic PBGD activity and restoring the physiological regulation of the heme synthesis pathway (Figure 2). Based on the PoC obtained in animal models, increasing PBGD levels appears to be a promising strategy for the etiological treatment of AIP, regardless of whether it is achieved by the administration of a recombinant rApoAI-PBGD protein[31], an rAAV-mediated GT[32], or mRNA encapsulated in LNPs[33]. Indeed, a single administration of the PBGD-mRNA induced a rapid and efficient overexpression of a functional PBGD protein in the liver of mice and large animals. The messenger RNA technology is being successfully tested in several clinical trials for five metabolic diseases (NCT 05095727: Glycogen storage disease Type 1A; NTC 04574830: Glycogen storage disease Type 3; NCT05130437: Propionic Acidemia; NCT 04899310: Methylmalonic Acidemia; NTC 04442347: Ornithine transcarbamilase deficiency) so that experience could quickly be transferred to patients with porphyria. Furthermore, this product could be applied to all patients regardless of clinical course, both chronic and sporadic presentations, as well as ASHE until normalization of the urinary excretion of porphyrin precursors.
This entry is adapted from the peer-reviewed paper 10.3390/life12111858
References
- Manceau, H.; Gouya, L.; Puy, H.; Acute Hepatic and Erythropoietic Porphyrias: From ALA Synthases 1 and 2 to New Molecular Bases and Treatments.. Curr. Opin. Hematol 2017, 24, 198-207, 10.1097/MOH.0000000000000330.
- Bissell, D.M.; Anderson, K.E.; Bonkovsky, H.L.; Porphyria. N. Engl. J. Med. 2017, 377, 862–872, 10.1056/NEJMra1608634.
- Balwani, M.; Erythropoietic Protoporphyria and X-Linked Protoporphyria: Pathophysiology, Genetics, Clinical Manifestations, and Management.. Mol. Genet. Metab. 2019, 128, 298–303, 10.1016/j.ymgme.2019.01.020.
- Chen, B.; Solis-Villa, C.; Hakenberg, J.; Qiao, W.; Srinivasan, R.R.; Yasuda, M.; Balwani, M.; Doheny, D.; Peter, I.; Chen, R.; et al. Acute Intermittent Porphyria: Predicted Pathogenicity of HMBS Variants Indicates Extremely Low Penetrance of the Auto-somal Dominant Disease. . Hum. Mutat. 2016, 37, 1215–1222, 10.1002/humu.23067.
- Unzu, C.; Sampedro, A.; Mauleón, I.; Vanrell, L.; Dubrot, J.; de Salamanca, R.E.; González-Aseguinolaza, G.; Melero, I.; Prie-to, J.; Fontanellas, A.; et al. Porphobilinogen Deaminase Over-Expression in Hepatocytes, but Not in Erythrocytes, Prevents Accumulation of Toxic Porphyrin Precursors in a Mouse Model of Acute Intermittent Porphyria. . J. Hepatol. 2010, 52, 417–424, 10.1016/j.jhep.2009.09.003.
- Lissing, M.; Nowak, G.; Adam, R.; Karam, V.; Boyd, A.; Gouya, L.; Meersseman, W.; Melum, E.; Ołdakowska-Jedynak, U.; Reiter, F.P.; et al. Liver Transplantation for Acute Intermittent Porphyria.. Liver Transplant. Off. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transplant. Soc. 2021, 27, 491–501, 10.1002/lt.25959.
- Dowman, J.K.; Gunson, B.K.; Bramhall, S.; Badminton, M.N.; Newsome, P.N.; Liver Transplantation from Donors with Acute Intermittent Porphyria. . Ann. Intern. Med. 2021, 154, 571–572, .
- Handschin, C.; Lin, J.; Rhee, J.; Peyer, A.-K.; Chin, S.; Wu, P.-H.; Meyer, U.A.; Nutritional Regulation of Hepatic Heme Biosynthesis and Porphyria through PGC-1α. . Cell 2005, 122, 505–515, 10.1016/J.CELL.2005.06.040.
- Schmitt, C.; Lenglet, H.; Yu, A.; Delaby, C.; Benecke, A.; Lefebvre, T.; Letteron, P.; Paradis, V.; Wahlin, S.; Sandberg, S.; et al. Recurrent Attacks of Acute Hepatic Porphyria: Major Role of the Chronic Inflammatory Response in the Liver. . J. Intern. Med 2018, 284, 78-91, 10.1111/joim.12750.
- Immenschuh, S.; Baumgart-Vogt, E.; Mueller, S.; Heme Oxygenase-1 and Iron in Liver Inflammation: A Complex Alliance. . Curr. Drug Targets 2010, 11, 1541–1550, 10.2174/1389450111009011541.
- Meyer, U.A.; Schuurmans, M.M.; Lindberg, R.L.P.; Acute Porphyrias: Pathogenesis of Neurological Manifestations. . Semin. Liver Dis. 1998, 18, 43-52, 10.1055/s-2007-1007139.
- Jaramillo-Calle, D.A.; Solano, J.M.; Rabinstein, A.A.; Bonkovsky, H.L.; Porphyria-Induced Posterior Reversible Encephalopathy Syndrome and Central Nervous System Dysfunction.. Mol. Genet. Metab. 2019, 128, 242–253, 10.1016/J.YMGME.2019.10.011.
- Petrides, P.E.; Therapy Follows Diagnosis: Old and New Approaches for the Treatment of Acute Porphyrias, What We Know and What We Should Know. . Diagnostics 2022, 12, 1618, 10.3390/diagnostics12071618.
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.C.; Gouya, L.; et al. Acute Intermittent Porphyria Causes Hepatic Mitochondrial Energetic Failure in a Mouse Model. . Int. J. Biochem. Cell Biol. 2014, 51, 93–101, 10.1016/j.biocel.2014.03.032.
- Solares, I.; Izquierdo-Sánchez, L.; Morales-Conejo, M.; Jericó, D.; Castelbón, F.J.; Córdoba, K.M.; Sampedro, A.; Lumbreras, C.; Moreno-Aliaga, M.J.; Enríquez de Salamanca, R.; et al. High Prevalence of Insulin Resistance in Asymptomatic Patients with Acute Intermittent Porphyria and Liver-Targeted In-sulin as a Novel Therapeutic Approach.. Biomedicines 2021, 9, 255, 10.3390/biomedicines9030255.
- Ardaiz, N.; Gomar, C.; Vasquez, M.; Tenesaca, S.; Fernandez-Sendin, M.; Di Trani, C.A.; Belsué, V.; Escalada, J.; Werner, U.; Tennagels, N.; et al. Insulin Fused to Apolipoprotein A-I Reduces Body Weight and Steatosis in DB/DB Mice. . Front. Pharmacol. 2020, 11, 591293, 10.3389/fphar.2020.591293.
- Song, P.; Kwon, Y.; Yea, K.; Moon, H.-Y.; Yoon, J.H.; Ghim, J.; Hyun, H.; Kim, D.; Koh, A.; Berggren, P.-O.; et al. Apolipoprotein A1 Increases Mitochondrial Biogenesis through AMP-Activated Protein Kinase.. Cell. Signal. 2015, 27, 1873–1881, 10.1016/j.cellsig.2015.05.003.
- Buendía-Martínez, J.; Barreda-Sánchez, M.; Rodríguez-Peña, L.; Ballesta-Martínez, M.J.; López-González, V.; Sánchez-Soler, M.J.; Serrano-Antón, A.T.; Pérez-Tomás, M.E.; Gil-Ferrer, R.; Avilés-Plaza, F.; et al. Health Impact of Acute Intermittent Porphyria in Latent and Non-Recurrent Attacks Patients. . Orphanet J. Rare Dis. 2021, 16, 106, 10.1016/j.cellsig.2015.05.003.
- Stölzel, U.; Doss, M.O.; Schuppan, D.; Clinical Guide and Update on Porphyrias. . Gastroenterology 2019, 157, 365–381.e4, 10.1053/j.gastro.2019.04.050.
- Anderson, K.E.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J.; Recommendations for the Diagnosis and Treatment of the Acute Porphyrias.. Ann. Intern. Med. 2005, 142, 439–450, 10.7326/0003-4819-142-6-200503150-00010.
- Gouya, L.; Ventura, P.; Balwani, M.; Bissell, D.M.; Rees, D.C.; Stölzel, U.; Phillips, J.D.; Kauppinen, R.; Langendonk, J.G.; Desnick, R.J.; et al. EXPLORE: A Prospective, Multinational, Natural History Study of Patients with Acute Hepatic Porphyria with Recurrent Attacks.. Hepatology 2020, 71, 1546–1558, 10.1002/hep.30936.
- Tchernitchko, D.; Tavernier, Q.; Lamoril, J.; Schmitt, C.; Talbi, N.; Lyoumi, S.; Robreau, A.-M.; Karim, Z.; Gouya, L.; Thervet, E.; et al. A Variant of Peptide Transporter 2 Predicts the Severity of Porphyria-Associated Kidney Disease. . . J. Am. Soc. Nephrol. 2017, 28, 1924–1932, 10.1681/ASN.2016080918.
- Lissing, M.; Vassiliou, D.; Floderus, Y.; Harper, P.; Bottai, M.; Kotopouli, M.; Hagström, H.; Sardh, E.; Wahlin, S.; Risk of Primary Liver Cancer in Acute Hepatic Porphyria Patients: A Matched Cohort Study of 1244 Individuals. . J. Intern. Med. 2022, 291, 824–836, 10.1111/joim.13463.
- Fontanellas, A.; Avila, M.A.; Hydroxymethylbilane Synthase (Aka Porphobilinogen Deaminase): A Novel Metabolic Tumor Suppressor Gene in Hepa-tocellular Carcinoma. . J. Hepatol. 2022, 77, 912–914, 2022.
- 44. Marsden, J.T.; Guppy, S.; Stein, P.; Cox, T.M.; Badminton, M.; Gardiner, T.; Barth, J.H.; Stewart, M.F.; Rees, D.C.; Audit of the Use of Regular Haem Arginate Infusions in Patients with Acute Porphyria to Prevent Recurrent Symptoms. . JIMD Rep. 2015, 22, 57–65, 10.1007/8904_2015_411.
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. . N. Engl. J. Med. 2020, 382, 2289–2301, 10.1056/NEJMoa1913147.
- Pallet, N.; Mami, I.; Schmitt, C.; Karim, Z.; François, A.; Rabant, M.; Nochy, D.; Gouya, L.; Deybach, J.-C.; Xu-Dubois, Y.; et al. High Prevalence of and Potential Mechanisms for Chronic Kidney Disease in Patients with Acute Intermittent Porphyria. . Kidney Int. 2015, 88, 386–395, 10.1038/ki.2015.97.
- To-Figueras, J.; Wijngaard, R.; García-Villoria, J.; Aarsand, A.K.; Aguilera, P.; Deulofeu, R.; Brunet, M.; Gómez-Gómez, À.; Pozo, O.J.; Sandberg, S.; et al. Dysregulation of Homocysteine Homeostasis in Acute Intermittent Porphyria Patients Receiving Heme Arginate or Giv-osiran.. J. Inherit. Metab. Dis. 2021, 44, 961–971, 10.1002/jimd.12391.
- Fontanellas, A.; Ávila, M.A.; Arranz, E.; Enríquez de Salamanca, R.; Morales-Conejo, M.; Acute Intermittent Porphyria, Givosiran, and Homocysteine. . J. Inherit. Metab. Dis. 2021, 44, 790–791, .
- Lazareth, H.; Poli, A.; Bignon, Y.; Mirmiran, A.; Rabant, M.; Cohen, R.; Schmitt, C.; Puy, H.; Karras, A.; Gouya, L.; et al. Renal Function Decline with Small Interfering RNA Silencing Aminolevulinic Acid Synthase 1 (ALAS1). . Kidney Int. Rep. 2021, 6, 1904–1911, 10.1016/j.ekir.2021.04.004.
- Córdoba, K.M.; Serrano-Mendioroz, I.; Jericó, D.; Merino, M.; Jiang, L.; Sampedro, A.; Alegre, M.; Corrales, F.; Garrido, M.J.; Martini, P.G. V; et al. Recombinant Porphobilinogen Deaminase Targeted to the Liver Corrects Enzymopenia in a Mouse Model of Acute Inter-mittent Porphyria. . Sci. Transl. Med. 2022, 14, eabc0700, 10.1126/scitranslmed.abc0700.
- Unzu, C.; Sampedro, A.; Mauleón, I.; Alegre, M.; Beattie, S.G.; De Salamanca, R.E.; Snapper, J.; Twisk, J.; Petry, H.; Gonzá-lez-Aseguinolaza, G.; et al. Sustained Enzymatic Correction by RAAV-Mediated Liver Gene Therapy Protects against Induced Motor Neuropathy in Acute Porphyria Mice. . Mol. Ther. 2011, 19, 243-250, 10.1038/mt.2010.210.
- Jiang, L.; Berraondo, P.; Jericó, D.; Guey, L.T.; Sampedro, A.; Frassetto, A.; Benenato, K.E.; Burke, K.; Santamaría, E.; Alegre, M.; et al. Systemic Messenger RNA as an Etiological Treatment for Acute Intermittent Porphyria. . Nat. Med. 2018, 24, 1899–1909, 10.1038/s41591-018-0199-z.