Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Genome-wide association studies (GWAS) have identified the PICALM (Phosphatidylinositol binding clathrin-assembly protein) gene as the most significant genetic susceptibility locus after APOE and BIN1. PICALM is a clathrin-adaptor protein that plays a critical role in clathrin-mediated endocytosis and autophagy.
- PICALM
- Alzheimer’s disease
- GWAS
- amyloid β
1. Physiopathology of Alzheimer’s Disease and Genetic Risk Factors
Alzheimer’s disease (AD) is the most common cause of dementia, manifesting itself in cognitive deficits. AD is characterized by two neuropathological hallmarks: extracellular deposition of fibrils made up of amyloid β (Aβ) peptides as senile plaques [1] and intracellular accumulation of hyperphosphorylated tau as neurofibrillary tangles (NFTs) [2] (for review, see [3]).
Aβ is produced by successive cleavages of β-amyloid precursor protein (APP), encoded by APP gene located in chromosome 21, by β-secretase (BACE1) and γ-secretase [4]. APP is cleaved by BACE1 at its N terminal domain to release soluble APP-β and the membrane-bound APP C-terminal fragment (β-CTF or C99). The γ-secretase is a complex of four protein subunits consisting of presenilin (PSEN), Nicastrin, presenilin enhancer (PEN) and APH1 (anterior pharynx-defective 1). Sequential cleavages of APP by BACE1 and γ-secretase lead to production of Aβ peptides of 38-, 40- and 42-amino acids in length. Aβ peptides are principally produced in endosomes and are released from neurons in a synaptic activity-dependent manner [5]. Aβ peptides adopt aggregation-prone conformations that are resistant to proteolysis and form oligomers, protofibrils and fibrils. Due to hydrophobicity at its C-terminus, Aβ42 has greater aggregation properties [6].
Tau is a microtubule-associated protein and has a role in stabilizing neuronal microtubules. Tau is encoded by MAPT (microtubule-associated protein tau) gene located on chromosome 17. Tau is natively soluble and unfolded, but tau can be aggregated into oligomers and fibrils in the presence of pathologically misfolded tau or polyanions [7]. In AD, tau aggregates form paired helical filaments (PHF) or straight filaments [8]. Compelling evidence suggests that tau pathology, defined by the accumulation of hyperphosphorylated aggregated forms of tau, is strongly associated with neurodegeneration and cognitive impairment in AD [6,9]. Tau pathology propagates between neuroanatomically connected areas throughout the brain during the progression of AD. The stereotypical aspect of tau pathology progression forms the basis of neuropathological Braak staging of tau pathology [10]. The stage of tau pathology better correlates with cognitive decline than Aβ load in AD patients [9]. Numerous studies have suggested that both synthetic and AD brain-derived tau fibrils can propagate in a prion-like manner by recruiting normal unfolded tau into pathological tau aggregates [11]. Tau aggregation can also be found in neurons and glia of other human neurological disorders, so-called tauopathies, such as frontotemporal lobar degeneration (FTLD) with tau pathology, progressive supranuclear palsy (PSP), Pick disease, corticobasal degeneration (CBD), etc. [12].
AD can be generally classified into two subgroups according to the age of onset. AD cases occurring earlier than age 65 are termed early-onset AD (EOAD), constituting less than 5% of all cases. Early-onset AD includes familial AD (FAD) inherited in an autosomal-dominant manner and can present at a very early age of onset and with a more rapid rate of disease progression, constituting 1–2% of AD cases. A number of FAD mutations have been identified in the genes of APP, Presenilin1 (PSEN1) and Presenilin2 (PSEN2) (γ-secretase component) [13].
Since age is the most important risk factor for AD [14], the great majority of cases, estimated at more than 95%, occur after age 65, constituting late-onset AD (LOAD). There was little knowledge of genetic risk factors for LOAD except for Apolipoprotein (APOE) risk allele APOE4 till the breakthrough made by genome-wide association studies (GWAS) [15,16]. To date, GWAS have identified about 75 single nucleotide polymorphisms (SNPs), including APOE4, BIN1, PICALM, CLU, CR1, ABCA7, TREM2, MS4A and APH1B, with a significant association for LOAD susceptibility [17]. These LOAD-susceptibility genes are functionally implicated in multiple cellular processes such as synaptic function, lipid metabolism, inflammation, endocytosis, cytoskeletal transports, and tau and amyloid pathways in AD pathologies [18,19,20]. Nevertheless, the mechanisms by which the GWAS hit genes modulate the AD risk remain largely elusive and have been under active scrutiny in the field of functional genomics in the post GWAS era. The vast majority of these variants are located in the noncoding region [21], and such variants are supposed to exert phenotypic effects via the perturbation of transcriptional gene promoters and enhancers [22].
2. Biology of PICALM
2.1. Tissue Expression, Cellular Functions and Protein Interaction of PICALM
The human PICALM gene is located on chromosome 11 [28]. There are 24 transcripts as splice variants, 15 of them coding protein [28], whose sizes vary from 134 to 652 amino acids [29], and the molecular weights of the major isoforms are estimated at 65–75 kDa due to alternative splicing of exon 13 [30]. PICALM is a ubiquitous protein and is expressed in the brain, muscle, kidney, urinary bladder, connective tissues, bone marrow, lymphoid tissues and in female tissues, including breast and placenta, https://www.proteinatlas.org/ENSG00000073921-PICALM/tissue (accessed on 1 June 2022). [31]. In the human brain, PICALM is abundantly expressed in the microglia, oligodendrocytes, endothelial cells, neurons, vascular mural and choroid plexus cells (for further information on the localization of PICALM in the AD brain, see 3.3.1) [30,32,33,34,35,36]. In the mouse brain, PICALM mRNA is highly expressed in the hippocampus [31], in particular in the dentate gyrus granule cells, as detected by a spatial transcriptomic study [37,38].
STRING database analysis revealed predicted protein–protein interactions of PICALM [39,40]. PICALM interacts with key proteins for clathrin-mediated endocytosis, intracellular trafficking and signaling (Figure 1). Interestingly, PICALM tightly interacts with other proteins encoded by GWAS hit genes for LOAD, such as BIN1, CD2AP, EPHA1, AP2A1 and AP2A2 [41]. PICALM is almost perfectly colocalized with the AP2 adaptor and AP2 α-adaptin subunits (encoded by AP2A1 and AP2A2) [42,43]. Interestingly, gene variants, splicing defects and altered expression of AP2A1 and AP2A2 have been recently shown to have a significant association with LOAD susceptibility [41]. Mounting evidence suggests PICALM has multiple roles in cellular functions and homeostasis as a clathrin adaptor for clathrin-mediated endocytosis, erythroid maturation, transferrin uptake, lipid homeostasis, autophagy, neuronal polarity, neuritic prolongation and synaptic vesicle turnover [44].
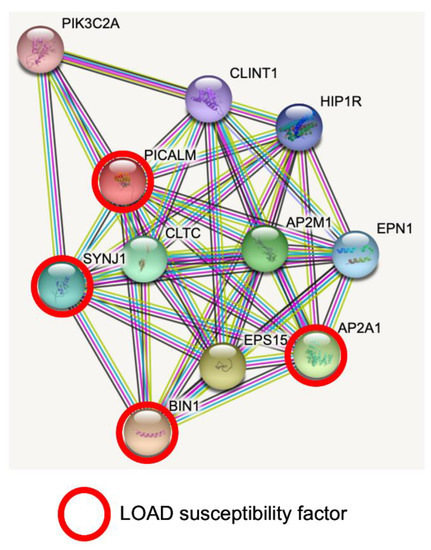
Figure 1. PICALM interacts directly with several GWAS hits identified in LOAD. Predicted protein–protein interactions by STRING database analysis (https://string-db.org (accessed on 1 June 2022)) show several key proteins for clathrin-mediated endocytosis and intracellular trafficking as PICALM interactors [40]. AP2A1 and BIN1 have been identified as GWAS hits [41]. SNPs of SYNJ1 have significant associations with disease onset of AD [45]. CLTC: Clathrin heavy chain 1. SYNJ1: Synaptojanin1. PIK3C2A: Phosphatidylinositol 4-phosphate 3-kinase C2 domain-containing subunit α. CLINT1: Clathrin interactor 1. HIP1R: Huntingtin-interacting protein 1-related protein. AP2M1: AP2 complex subunit mu. EPN1: Epsin-1. AP2A1: AP2 complex subunit α-1. EPS15: Epidermal growth factor receptor substrate 15. BIN1: Myc box-dependent-interacting protein 1.
2.1.1. PICALM and Clathrin-Mediated Endocytosis
Clathrin-mediated endocytosis, also called receptor-mediated endocytosis, is a process leading to the internalization of plasma membrane-associated cargo molecules and subsequent trafficking through intracellular vesicle compartments. PICALM is necessary for clathrin assembly at the plasma membrane by binding to AP2 and clathrin (Figure 2A). PICALM interacts simultaneously with AP1 and clathrin at the trans-Golgi network in HeLa cells [43]. Several independent studies have reported that the depletion of PICALM leads to an increase in the size of clathrin-coated vesicles (CCVs) and to an increase in heterogeneity in the CCVs shape in cultured cells of rat hippocampus [46] and of HeLa cells [43,47]. CRISPR/Cas9-mediated PICALM disruption also leads to an increase in the number of early endosomes in HeLa cells [48]. A similar observation was obtained by the depletion of uncoordinated protein-11 (unc-11), the ortholog of PICALM in neurons of C. elegans [49]. Another study suggests that PICALM is required for efficient clathrin coat maturation in fibroblasts [50].
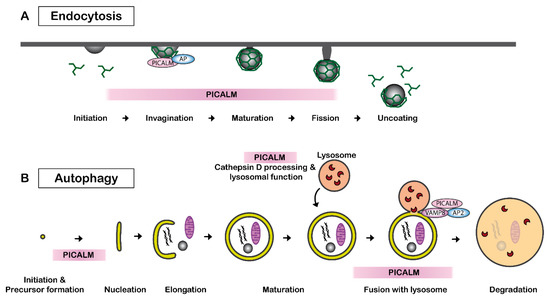
Figure 2. PICALM regulates clathrin-mediated endocytosis and autophagy. (A) PICALM is involved in clathrin-mediated endocytosis (CME) as a clathrin adaptor by facilitating the formation of clathrin-coated vesicles (CCVs). The interaction between PICALM, adaptor proteins (AP) and clathrin is critical to maintaining the form and sizes of CCVs. PICALM interacts with AP2 at the plasma membrane and AP 1 in the trans-Golgi network to form CCVs. (B) PICALM is also involved in multiple steps of autophagy process. PICALM is involved in the autophagy precursor formation at the autophagosome-lysosome fusion [51], maturation of Cathepsin D and lysosomal function [48].
2.1.2. PICALM, Hematopoiesis, Transferrin-Uptake and Cholesterol Homeostasis
Chromosomal translocations of PICALM have been observed in patients with acute lymphoblastic leukemia and acute myeloid leukemia [52]. In mice, nonsense point mutations of PICALM cause abnormal iron distribution, growth retardation, shortened life span and hematopoietic abnormalities [53].
PICALM is a critical protein, and homozygous PICALM knockout mice are dwarfed and die within 1 month after birth due to a deficiency in erythroid maturation and transferrin uptake [54]. There are controversies in the literature on the effects of PICALM on transferrin uptake. While some studies suggest that PICALM overexpression or down-expression did not alter transferrin uptake in HeLa cells or HEK293 cells [55,56], others observed a significant effect on transferrin uptake in the liver, bone marrow and erythroid cells from PICALM-deficient mice [50,54]. It is thus speculated that the deletion of PICALM may be partially compensated by other endocytic proteins in certain types of cells. Indeed, PICALM is dispensable in myeloid and B-lymphoid development [50].
Additionally, PICALM also modulates cellular cholesterol homeostasis, as PICALM expression influences the expression of genes encoding proteins involved in cholesterol biosynthesis and lipoprotein uptake [57]. Knockdown of PICALM leads to up to a 50% increase in cholesterol biosynthesis genes in in HEK293, murine embryonic fibroblasts (MEF) and Cath-a-differentiated (CAD) neuronal cells [57].
2.1.3. The Roles of PICALM in Neuronal Polarity and Synaptic Vesicle Sorting
PICALM and its neuronal homolog assembly protein 180 (AP180) have quite interesting functions in neuronal cells. Endocytosis is reduced in PICALM- or AP180-deficient neurons [58]. Interestingly, PICALM and AP180 have critical roles in establishing the polarity and neuritic growth in neuronal cells. While treatments with siRNA targeting PICALM lead to dendritic dystrophy, treatments with siRNA targeting AP180 abolish axonal extension in rat primary neuronal culture. In contrast, neurons overexpressing AP180 or PICALM generate multiple axons [58].
PICALM is expressed in neurons and is present in synapses [36]. PICALM regulates membrane fusion by binding to R-SNAREs (soluble NSF attachment protein receptors) such as VAMP2, VAMP3 and VAMP8 [59,60]. Furthermore, PICALM regulates the internalization of VAMP2 (Synaptobrevin 2), the most abundant synaptic vesicle protein and a major R-SNARE component [56]. Since the AP180 N-terminal homology (ANTH) domain (see chapter 2.2) of PICALM interacts with VAMP2, both PICALM and AP180 regulate endocytic sorting of synaptic vesicles at active zones for neurotransmission [59]. PICALM and related ANTH-domain-containing proteins modulate the surface levels of calcium-permeable AMPA-type glutamate receptors (AMPARs) that mediate fast excitatory neurotransmission and excitotoxicity [61]. PICALM is thereby supposed to play roles in synaptic plasticity and learning by modulating both long-term potentiation (LTP) and long-term depression (LTD) [62].
2.1.4. PICALM and Autophagy
PICALM plays a key role in multiple steps in autophagy (Figure 2B). PICALM regulates the endocytosis of VAMP2, VAMP3 and VAMP8, which are essential for the autophagy process [60]. PICALM knockdown leads to a reduced fusion of autophagosome precursor vesicles, which is a VAMP2- and VAMP3-dependent process, and thus decreases autophagosome biogenesis. PICALM knockdown also influences the process of autophagosome-lysosome fusion, which is a VAMP8-dependent process [63]. Therefore, PICALM reduction results in a general reduction in autophagic flux in HeLa, HEK, CAD and MEF cells [51]. Additionally, PICALM is also implicated in the maturation of Cathepsin D, which is a critical lysosomal component [48]. Disruption of exon1 of PICALM leads to deficits in the maturation of Cathepsin D and autophagy in HeLa cells [48].
2.2. Protein Structure of PICALM
PICALM is constituted of an N-terminal ANTH-domain binding phosphatidyl inositol 4,5 bisphosphate (PtdIns(4,5)P2) and a C-terminal clathrin adaptor domain (Figure 3A) [42,64].
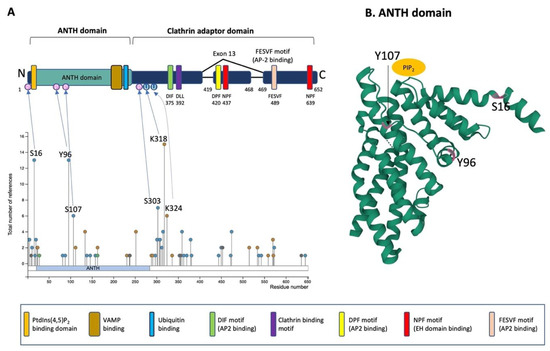
Figure 3. Schematic illustration of PICALM structure and post-translational modifications. (A) Schematic structure of PICALM. PICALM is constituted of two distinct domains: the ANTH (AP180 N-terminal homology) domain and clathrin adaptor domain. ANTH domain contains a PtdIns(4,5)P2 binding domain and vesicle-associated membrane protein (VAMP) binding domain. Clathrin adaptor domain contains several motifs that are critical to interact with endocytic protein, including DIF, DLL Clathrin binding, DPF, NPF and FESVF motifs. DIF motif binds to AP2. DLL motif is conserved and has weak affinity to Clathrin DPF (Aspartic acid-Proline-Phenylalanine) motifs. NPF (Asparagine-Proline-Phenylalanine) motif binds to AP2 and EH domains (Eps15 Homology domain). Four phosphorylation sites (S16, Y96, S107 and S303) and two ubiquitination sites (K318 and K324) have been described in more than 5 references and validated by mass-spectrometry and thus are highlighted [65]. Altogether, the reported PTMs of PICALM are 41 phosphorylation (S2, S5, T11, S16, T18, S20, S23, T82, Y88, Y96, S107, Y138, T146, T158, Y237, S297, T301, S303, S307, S308, T312, S315, S353, T355, T356, S359, T363, T370, T379, S381, S444, S450, S453, T472, S474, K515, K534, S537, S543, T573 and S645), 15 ubiquitination (K24, K112, K134, K163, K231, K252, K288, K291, K318, K324, K336, K515, K559, K570 and K571), 1 acetylation (K28), 1 mono-methylation (R9), 1 di-methylation (R636) and 1 sumoylation (K238). (B) Computational modeling of the structure of PICALM N-terminal ANTH domain is shown (https://www.rcsb.org/structure/3ZYM (accessed on 1 June 2022)) [66]. PICALM ANTH domain is constituted of 11 α-helices. Three known phosphorylation sites (S16, Y96 and S107) in ANTH domain are highlighted in pink.
The ANTH domain is a membrane-binding domain with specificity for phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) and acts as a universal adaptor for nucleation of clathrin coats. In silico modeling of N-terminal ANTH domain suggests a structure consisting of 11 α-helices [60] (Figure 3B). The PtdIns(4,5)P2 binding residues (Kx9Kx(K/R)(H/Y) are present on several helices. The PtdIns(4,5)P2 binding sites contain evolutionally conserved basic amino acid residues (K28, K38, K40 and K41, numbering according to the PICALM isoform of 652 amino acids) forming a positively charged patch on the surface to interact with phosphates of PtdIns(4,5)P2 [64]. The ANTH domain also contains the binding region for R-SNARE proteins, and PICALM can bind simultaneously PtdIns(4,5)P2 and VAMP8 [60]. PICALM interacts with R-SNARE proteins via its hydrophobic amino acid residues such as L219, F240, M244, I272 and L251 [60]. A recent study has shown that the ANTH domain directly binds to ubiquitin, and the ANTH domain can act as an adaptor for ubiquitinated protein [67].
In the C-terminal clathrin adaptor domain of PICALM contains critical motifs that allow PICALM to interact with clathrin and other endocytic machinery proteins. The clathrin binding motif is DLLDLQ (392–397 in the isoform of 652 amino acids). AP2-binding motifs are DIF (D375, I376 and F377), DPF (D420, P421 and F422) and FESVF (F489, E490, S491, V492 and F493). The asparagine-proline-phenylalanine (NPF) motif binds to Eps15 homology (EH) domains found in proteins associated with endocytosis and vesicle trafficking. Since exon 13 contains one DPF (AP2 binding) and one NPF (EH domain binding) motif, the splice variant without exon 13 is supposed to have reduced interaction with AP2 and other protein partners owning EH-domains [43].
2.3. Post-Translational Modifications of PICALM
PICALM undergoes several post-translational modifications (PTM), such as phosphorylation, methylation, ubiquitination, sumoylation and acetylation [65,66,68,69,70,71,72,73,74]. These post-translational modifications have been identified on numerous amino acid residues of PICALM: 41 phosphorylation, 15 ubiquitination, 1 acetylation, 1 mono-methylation, 1 di-methylation and 1 sumoylation sites have been described so far. Certain factors are known to induce some of the post-translational modifications on PICALM, such as phosphorylation by Aβ treatment in primary neuronal culture [75], by BI2536, a selective inhibitor of protein kinase Polo-Like Kinase 1 (PLK1), inducing phosphorylation (Y138, T146 and S537) [76], by ischemia (S303 and S315) [77] and by nocodazole, an inhibitor of microtubule polymerization (S474 and S645) [78]. The most well-characterized PTM of PICALM are the four phosphorylation sites (S16, Y96, S107 and S303) and two ubiquitination sites (K318 and K324) that have been validated by mass-spectrometry and have been described for more than 5 times [65]. While the phosphorylation on the ANTH domain may influence the interactions of PICALM with PtdIns(4,5)P2 or R-SNARE proteins, the other phosphorylation residues are located in the C-terminal clathrin adaptor domain exhibiting pivotal functions in protein–protein interaction of PICALM with clathrin and other endocytic machinery proteins. Therefore, it is speculated that phosphorylation of PICALM should play a role in regulating interaction with PtdIns(4,5)P2 and other protein binding partners. For instance, the R-SNARE binding domain contains several sites that can undergo PTM, such as one phosphorylation (Y237), two ubiquitination (K231 and K252) and one sumoylation (K238), and thus, these PTM may regulate the interaction between PICALM and R-SNARE.
PICALM is a substrate of calpain and caspase [33,79,80]. Nevertheless, it remains largely unknown if the phosphorylation and ubiquitination of PICALM affect its protein stability or degradation.
Given that PICALM contains more than 130 putative phosphorylation sites, PICALM phosphorylation may be dysregulated in AD brains not only in terms of its global phosphorylation level but also in the pattern of site-specific phosphorylation. Indeed, Aβ exposure increases PICALM phosphorylation in primary rat neuronal cortical culture [75] though phosphorylation sites and the involved kinases remain to be identified. While it is speculated that phosphorylation may change the interaction of PICALM with its binding partners such as R-SNAREs, PtdIns(4,5)P2 or clathrin, it remains elusive which kinases are involved in PICALM phosphorylation, whether PICALM phosphorylation is misregulated during the progression of AD, whether PICALM phosphorylation has any impact on its protein stability/degradation or its subcellular localization and whether phosphorylation of PICALM may cause a loss of function and/or a gain of toxicity.
This entry is adapted from the peer-reviewed paper 10.3390/cells11243994
This entry is offline, you can click here to edit this entry!