Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Ulcerative colitis (UC) and Crohn's disease (CD) have been recognized as occidentalized diseases, due to their higher rates of incidence and prevalence in occidental countries. CD mostly presents between 20 and 30 years, whereas UC mostly presents between 30 and 40 years, as well as from 60 to 70 years.
- Crohn’s disease
- ulcerative colitis
- serotonin
- bowel disease,
1. Risk Factors and Clinical Manifestations
Ulcerative colitis (UC) and Crohn's disease (CD) have been considered as multi-factorial pathologies, including factors such as antibiotic use, viral and bacterial infections, processed and sugary foods, and alteration in the intestinal microbiota (intestinal dysbiosis) which have been related to an increased risk of inflammatory bowel disease (IBD) [8,110,111,112]. Notably, smoking has been described as having a controversial effect on IBD, as it acts as a risk factor in CD due to the production of free radicals, thus perpetuating inflammation; meanwhile, in UC, smoking has been identified as playing a protective role [3,8]. IBD is associated with factors such as abdominal delivery (CD, OR = 1.38, 95% CI: 1.12–1.70; UC, OR = 1.08, 95% CI: 0.87–1.33), antibiotics exposure (CD, OR = 1.74, 95% CI: 1.35–2.23; UC, OR = 1.08, 95% CI: 0.91–1.27), and sucrose ingestion (CD, RR = 1.09, 95% CI: 1.02–1.16; UC, RR = 1.10, 95% CI: 1.02–1.18), among others [8]. The dietary composition has been also considered as a risk factor, as it has the capacity to disrupt the normal gut microbiota, especially when foods such as sodas, chocolate, and artificial sweeteners are included [5]. Intestinal permeability has been found to be increased in mice fed a high-sugar diet [113]. Besides the influence of diet, increased serum LPS levels and decreased microbiota diversity can lead to reduced production of SCFAs [114].
CD is characterized by transmural damage. Patients may present with perianal pain, bleeding, incontinence, fistulization, abscesses, and hemorrhoidal illness [115]. CD can also be characterized by the presence of extraintestinal manifestations, the most common being enthesitis and axial or peripheral arthritis [116,117]. On the other hand, UC can produce chronic inflammation of the colonic mucosa, leading to manifestations such as proctitis, bloody stools, abdominal pain, fatigue, fecal incontinence, arthralgia, and erythema nodosum [118]. In both UC and CD, there is also involvement of the central nervous system (CNS), specifically leading to psychological or psychiatric manifestations [119]. It has been observed that between 15% and 25% of patients with IBD developed depression, while 30% presented with anxiety [120]. Furthermore, it may be accompanied by sleep difficulties and fatigue [121]. As part of the neurological involvement in patients with IBD, deficits in attention and executive function in adults have been observed [122].
Genetic Susceptibility and Inflammatory Bowel Disease
IBD, similarly to other inflammatory diseases such as autoimmune diseases, has been related to different genes; although IBD has not been recognized as a hereditary disease, several articles provide information about increased susceptibility, disease activity and treatment response related with these genes and their genetic variants (Table 1). As an example, in a trans-ancestry association study, in European, East Asian, Indian and Iranian populations, several risk loci for IBD were found. These loci included NOD2, ATG16L1, and IL-23R [123]. NOD2 gene variants such as p.Arg702Trp were able to provide an increased risk of IBD [14,124]. The rs2241880 gene variant of ATG16L1 has also been closely related with the maintenance of human intestinal cell homeostasis and autophagy processes in patients with IBD [16,125]. In an interesting review by Nour Younis et al., a wide range of genes related to IBD, together with their reported genetic variants, were considered [13]. Based on wide-genome studies, gene variants in genes such as ATG16L1 (rs2241880: OR = 0.74; 95% CI: 0.65–0.84; p < 0.001), PTPN2 (Protein tyrosine phosphatase non-receptor type 2) and of IL-23R (rs11209026 allele A; OR = 0.32; 95% CI: 0.17–0.60; p < 0.001) were related to increased susceptibility to IBD, and even to disease course and treatment outcomes [13].
Table 1. Genes related to Inflammatory Bowel Disease.
| Gene | Locus | Effect | Reference |
|---|---|---|---|
| NOD2 | 16q12.1 | IBD increased risk | [13,14,123,126] |
| ATG16L1 | 2q37.1 | Impaired intracellular bacteria clearance in IBD, intestinal autophagy | [13,16,127,128] |
| PTPN2 | 18p11.21 | IBD increased risk | [13,129,130] |
| IL-23R | 1p31.3 | IBD susceptibility, Crohn’s disease risk | [13,15,131] |
| IL-10 | 1q32.1 | IBD steroid dependency, early onset IBD | [13,132,133] |
| HNF4α | 20q13.12 | IBD susceptibility | [13,18,134] |
NOD2: Nucleotide-binding oligomerization domain-containing protein 2; ATG16L1: Autophagy-related 16-like 1 protein; PTPN2: Protein tyrosine phosphatase non-receptor type 2; IL-23R: Interleukin 23 receptor; IL-10: Interleukin 10; HNF4α: Hepatocyte Nuclear Factor 4 alpha.
2. Intestinal Barrier Disruption and Over-Activated Immune Response in Inflammatory Bowel Disease
The intestinal epithelial barrier, together with the intestinal microbiota, are considered an elemental functional unit in the physiology and pathophysiology of the GI. As part of the events that contribute to the pathogenesis of IBD, an alteration in the structure of the intestinal barrier can lead to an altered immune response and intestinal dysbiosis. This phenomenon has been clearly established in pathogen-free mice, where an alteration in the intestinal epithelial cells was observed, together with several abnormalities in the microvilli and a decrease in cellular renovation of the gut (Figure 1) [135]. TJs, such as zonula occludens 1 (ZO-1) and zonula occludens 2 (ZO-2), can be influenced and affected by the intestinal microbiota. TJs can be regulated by bacteria, such as Lactobacillus rhamnosus, Acidophilus plantarum, and Bifidobacterium infantis, through the activation of TLRs, causing increases in the expression of claudin 3, ZO-1, and claudin 4 [136]. In patients with a previous established diagnosis of IBD, an increased paracellular permeability has been observed in almost 40% of patients with CD [137].
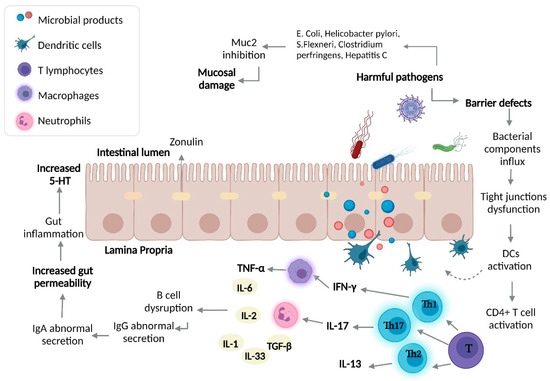
Figure 1. Intestinal dysbiosis and immune response in IBD. The over-growth of harmful bacteria and gut barrier defects cause an influx of bacterial components, leading to activation of the immune response (both innate and adaptive). In UC, Th2 differentiation prevails, while in CD, a Th1 and Th17 adaptive immune response prevails. The influx of bacterial components, adaptive immune response, and harmful bacterial over-growth lead to increased gut permeability, resulting in increased gut 5-HT causing an inflammatory process. DCs, dendritic cells; IFN-γ, interferon-γ; IL-17, interleukin 17; IL-13, interleukin 13; TNF-α, tumor-necrosis factor α.
Maintenance of the intestinal epithelial integrity depends on the mucus layer; nevertheless, Akkermansia muciniphila, Ruminococcus spp., Enterococcus, Bifidobacterium spp., and Bacteroides can degrade mucin and favor the colonization of harmful bacteria [138]. In patients with IBD, increases in Ruminococcus gnavus, Rumminococcus torques, Bacteroides fragilis, and Bacteroides vulgatus, containing mucolytic enzymes such as α-galactosidase, sulphatase, neuraminidase, and β-galactosidase, have been observed [139]. An alteration in the relation between Bacteroidetes, Firmicutes, and Actinobacteria, as well as a decrease in Proteobacteria and an increase in new bacterial groups, promotes an alteration to homeostasis in a process called intestinal dysbiosis. The equilibrium between host micro-organisms (also called symbiosis) can be affected by nutritive factors, fats, carbohydrates, and drug or antibiotic abuse, among others [110]. Bacteria such as E. Coli, Klebsiella spp., Proteus, Enterobacter, Shigella spp., Salmonella spp., and Serratia have been studied as pathogen micro-organisms which are capable of inducing inflammation and intestinal manifestations [135,136].
A decrease has been observed in the fecal concentrations of Bacteroides fragilis and B. vulgatus, both of which have protective potential, where their absence could lead to perpetuated inflammation and the development of IBD [137]. Reductions in the levels of Firmicutes and Proteobacteria were found to be the most reported and consistent changes in patients with IBD. Meanwhile, a metagenomic analysis reported an increase in enterobacteria, most commonly E. coli [136,138,140]. F. prausnitzii possesses anti-inflammatory properties; however, it has been shown to be decreased in patients with IBD (specifically, CD) [141,142]. Some probiotics are able to reinforce the intestinal barrier through the production of defensins and zonula occludens 2 proteins [143]. On the other hand, in patients with IBD, an increase in Malassezia restricta—a fungus generally found in the skin, which is able to promote the production of pro-inflammatory cytokines by immune cells—has been observed, specifically in those who were described as having a mutation in the CARD9 gene, which has been described in IBD [144].
IBD patients with over-expression of carcinoembryonic antigen cell adhesion molecule 6 (CEACAM 6) are more susceptible to EIEC infection, due to the ability of E. coli to bind to CEACAM6 [136]. EIEC has the potential to promote the production of TNF-α by macrophages and survive inside them, thanks to genes such as ATG16L1, immunity-related GTPase family protein (IRGM), and NOD2. When these genes suffer some mutation, these capacities disappear and the EIEC proteins (FimH) are able to bind to TLR4, generating, as a consequence, an inflammatory response [137]. Furthermore, a relation between serotonin, SERT, and TLR-2 has been observed: Ahmad Qasem et al. found that, after infecting Caco-2 cells with Mycobacteria paratuberculosis (MAP), there was an increase in the levels of pro-inflammatory cytokines and TLR2 and, consequently, due to the stimulation of this pro-inflammatory cascade, decreased SERT and IL-10 expression [145].
Due to the linkage between intestinal dysbiosis and GBA, several in vitro and in vivo models have demonstrated the important influence of certain pathogenic bacteria present in patients with IBD and/or the beneficial effect of specific bacteria over SERT and serotonin signaling, as detailed in Table 2.
Table 2. Relationships between bacteria and SERT function.
| Bacteria | Mechanism | Model | References |
|---|---|---|---|
| Enteropathogenic E. coli | Activation of protein tyrosine phosphatase, a process that leads to SERT inhibition | Caco-2 cells infected with E. coli | [146,147] |
| Listeria monocytogenes |
Reduced SERT expression related to a transcriptional change in TLR10 and TLR2 | Caco-2/TC7 cells infected with Listeria monocytogenes | [148] |
| Akkermansia muciniphila |
Interaction between activated TLR2 and SERT causes reduced SERT expression | Caco-2 cells infected with Akkermansia muciniphila | [149] |
| Lactobacillus acidophilus |
Up-regulation of SERT mRNA | Lactobacillus acidophilus and B. longum interaction with HT-29 and Caco-2 cells | [150] |
| Lactobacillus rhamnosus |
SERT Gene and protein up-regulation | Wistar rats implemented with probiotics and prebiotics | [151] |
| Campylobacter jejuni | EC hyperplasia and reduced SERT expression | C57BL/6 mice infected with T. Spiralis and C. jejuni | [152] |
| Salmonella typhimurium |
Inhibition of SERT by TLR4 activation | Mice and Caco-2 cells infected with S. typhimurium | [153,154] |
2.1. Immune Over-Activation in IBD
Innate lymphoid cells (ILCs) come from a common lymphoid progenitor [155] and are differentiated into Natural Killer cells (NK), innate lymphoid cells 1 (ILC1s), innate lymphoid cells 2 (ILC2s), and innate lymphoid cells 3 (ILC3s) [143]. When ILC1s receive stimuli through IL-12, IL-15, and IL-18, there is a release of interferon gamma (IFN-γ), which promotes the ability of macrophages and DCs to remove intracellular bacteria-presenting antigens through the expression of major histocompatibility complex (MHC) and adhesion molecules. In contrast to ILC1s, ILC2s have the ability to release IL-5, IL-9, and IL-13 under certain stimuli, while ILC3s are producers of IL-22 and IL-17 [140]. An alteration in ILC1s and ILC3s in IBD has been described, as IL-12 is able to induce the differentiation of ILC3s into ILC1S to produce IFN-γ; furthermore, ILC3s can pass into a process of maturation when interacting with the microbiota. Pathogen-free mice with decreased IL-22 level presented with alterations, which led to disruption of the intestinal symbiosis [156]. Furthermore, the interaction of IFN-γ with ILCs promotes the migration of neutrophils, lymphocytes, and macrophages, as well as the activation of endothelial cells, thus causing a disruption in the intestinal barrier by affecting TJs [138].
Besides the importance of ILCs, cells such as macrophages are able to act as a bridge between the innate and adaptive immune response. Macrophages are susceptible to a process called polarization, which allows for the differentiation of macrophages into M1 and M2 sub-types, depending on the received stimuli. M1 macrophages are able to trigger an inflammatory response through the production of pro-inflammatory biomarkers such as IL6, IL-12, and TNF-α, while M2 macrophages possess anti-inflammatory properties [139]. In patients with IBD, an increase in the levels of IL-33 and hyperplasia of caliciform cells has been observed, accompanied by macrophage M2 polarization [157]. In particular, in macrophages of patients with IBD, the intracellular replication of bacteria including E. coli, Micobacterium, Salmonella, Shigella, Coxiella, Brucella, Legionella, and Listeria has been reported [158].
Together with the macrophages, neutrophils are the most abundant innate immune cell (approximately 70%). In IBD, these cells are responsible for the increased production of ROS, which causes damage to the intestinal epithelial barrier and can activate an inflammatory cascade [127]. Neutrophils are capable of forming a special defense mechanism—Neutrophil Extracellular Traps (NETs)—which are responsible for catching the pathogen component in a microbicidal environment, guaranteeing regulation of the immune response and a highly efficient defense mechanism [159]. Neutrophils migrate to the area of inflammation through interaction with components such as selectins and intracellular/vascular adhesion molecules (e.g., ICAM-1 and VCAM-1) [160]. One of the main findings in histopathological samples of patients with IBD was neutrophil infiltration with the presence of citrullinated histone H3 (citH3) and some other specific NETs products; this formation of NETs in patients with IBD can be considered a result of release and stimulation by TNF-α [161].
Together with macrophages and neutrophils, DCs play an important role in maintaining immune tolerance, considering the effects of nutrients and commensal bacteria [162]. In mucosal samples of patients with IBD, a decrease in sub-populations of CD103+ has been observed, as well as an increase in the expression of TLRs, thus generating an increase in immune responses and leading to a loss of immune tolerance [163]. In CD, a imbalance has been documented in the DCs, which may contribute to an excessive T cell response; in samples from patients with CD, a high expression of TLRs (TLR4) was also observed, as well as an increase in CD11c+, which produces IL-12 and IL-16 [164].
Once the innate immune response is over, the adaptive immunity is activated. This type of immunity is characterized by the generation of memory, with the capacity to confer long-term immunity, mediated by T and B lymphocytes [165].
Th1 Response and Crohn’s Disease
An interplay between the microbiota, immune system, and IBD has been described in terms of decreases in Bacteroides and Firmicutes, along with increases in Clostridium, Gammaproteobacteria, Actinobacteria, enteroinvasive Escherichia coli (EIEC), and ILC1s, with high expression of IL-17A, IL-22, and IL-23 receptor (IL-23R) [166].
CD4 T helper lymphocytes differentiate to Th1, Th2, Treg, Th17, TFH, or Th9 under specific stimuli; notably, a Th1 and Th17 response has been observed as part of the pathophysiology of CD [166]. As previously mentioned, T lymphocytes can differentiate under various chemical stimuli into Th1 lymphocytes, which are producers of IFN-γ, IL-12, IL-17, and IFN-γ [167]. The IL-17/IL-23 axis is a key actor in CD: when IL-23 binds to its receptor, IL-23R, which is expressed in cells including DCs, macrophages, neutrophils, NK cells and ILCs [168], the activation of a kinase (jak2) and a tyrosin kinase (tyk2) causes phosphorylation of the receptor and the transcription 3 activator (STAT3) [169]. Single nucleotide variants (SNVs) reported in the IL-23R gene in chromosome 1p31 (rs10889677) lead to increased IL-23R level, favoring chronic inflammation in CD [168]. Furthermore, there exists a relationship between IL-23 and Th17 activation, with subsequent accumulation of IL-17 producer cells in patients with CD [169].
Th2 Response and Ulcerative Colitis
T lymphocytes differentiate to Th2 lymphocytes after stimulation by IL-4, IL-33, and a transcription factor (GATA3), leading to the final production of IL-4 and IL-13 [170]. Another effect that has been related to IL-13 is the damage that it can impose upon the intestinal epithelial barrier, as IL-13 is able to increase apoptosis in epithelial cells as well as induce disruptions in cellular unions such as claudins-2 [171]. It has been suggested that IL-13 is capable of activating a pro-apoptotic molecule—caspase 3 [172].
3. Serotonin and Gut–Brain Axis Dysfunction in IBD
Due to the link between intestinal dysbiosis and GBA, it has been considered that there is an important influence of certain pathogenic bacteria present in patients with IBD and/or a beneficial effect of specific bacteria over SERT and serotonin signaling, as detailed in Table 2.
The GBA coordinates the release of the adrenocorticotropic hormone under stress, which can cause increases in intestinal permeability and glucocorticoid secretion [173]. The increased intestinal permeability associated with a high level of stress favors communication between the microbiota and nervous system [174,175]. Being a complex network, it has an influence over the neuroplasticity of the ENS during inflammation, leading to structural changes, including degradation and loss of enteric ganglion cells, causing an alteration in the normal neurotransmission and, therefore, gastrointestinal mechanosensitive alterations [176]. In view of the pro-inflammatory profile described by IL-1β, IL-6, and TNF-α, among others, leading to inhibition of the vagus nerve and activation of HPA axis (Figure 2) [177], S Haub et al. found that gut inflammation and a lack of IL-10 and SERT lead to abnormal serotonin signaling [178]. Besides the influence of serotonin and SERT over the maintenance of homeostasis in the GI tract, SERT acts as a determinant of the maintenance of bone mass in patients with IBD. Interestingly, osteoporosis is frequent in patients with IBD [179]. B. Lavoie et al. have demonstrated that, in mice, a lack of SERT due to induced colitis led to an incredible loss of trabecular bone mass. It has been found that the serotonin secreted by EC cells acts as a negative regulator of bone density through the inhibition of osteoblasts, which is a cell type responsible for producing and remodeling bone mass [180].
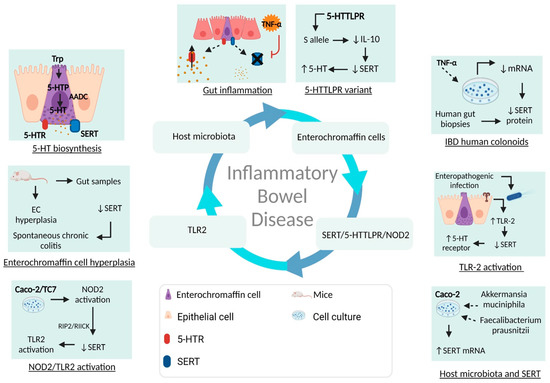
Figure 2. Overview of SERT, gut microbiota, and IBD pathogenesis. Serotonin is a monoamine exerting a wide range of biological effects through the intestinal microbiota, serotonin receptors, and SERT. After the biosynthesis of serotonin in enterochromaffin cells, there is the possibility that, in the context of gut inflammation, SERT may be affected by different pro-inflammatory mediators and bacteria, leading to dysregulation of mRNA, SERT proteins, and serotonin bioavailability. Trp, tryptophan; 5-HTP, 5-hydroxytryptophan; NOD2, Nucleotide oligomerization domain 2; TLR-2, Toll-like receptor 2; EC, enterochromaffin cells.
This entry is adapted from the peer-reviewed paper 10.3390/ijms232415632
This entry is offline, you can click here to edit this entry!