Metastatic spread represents the leading cause of disease-related mortality among cancer patients. Many cancer patients suffer from metastatic relapse years or even decades after radical surgery for the primary tumor. This clinical phenomenon is explained by the early dissemination of cancer cells followed by a long period of dormancy. Although dormancy could be viewed as a window of opportunity for therapeutic interventions, dormant disseminated cancer cells and micrometastases, as well as emergent outgrowing macrometastases, exhibit a generalized, innate resistance to chemotherapy and even immunotherapy. This therapeutic pan-resistance, on top of other adaptive responses to targeted agents such as acquired mutations and lineage plasticity, underpins the current difficulties in eradicating cancer. In the present review, we attempt to provide a framework to understand the underlying biology of this major issue.
- metastasis
- disseminated tumor cells
- E-cadherin
- therapy resistance
- dormancy
- metastatic microenvironment
- immune checkpoint blockade
- epigenetics
- metabolic plasticity
1. Introduction
Despite the major advances in cancer treatment in recent decades, most of the progress has been achieved through early diagnosis and treatment of pre-metastatic cancer. Unfortunately, metastatic disease remains essentially incurable for most cancers, as metastases are not amenable to removal due to wide dissemination, exhibit intrinsic generalized resistance to chemotherapy and immunotherapy, and rapidly develop acquired resistance to targeted therapy via adaptive mutations [1]. It is imperative to understand the biology of this generalized resistance in order to translate this understanding into novel therapeutic approaches that will improve patient outcomes.
The generalized resistance of metastases to chemotherapy is most clearly seen with neoadjuvant breast cancer treatment (systemically administered drugs before breast cancer surgery). Despite the fact that neoadjuvant chemotherapy will shrink the breast tumor, allowing a breast-conserving surgery and oftentimes leading to a complete pathological response, this fails to reflect on clinical outcomes, such as event-free survival and overall survival [2,3,4,5]. Thus, disseminated tumor cells (DTCs) that have left the primary tumor before resection often appear not to be eradicated by therapy but instead are intrinsically resistant. This differential therapeutic responsiveness between metastases and primary tumors may be attributed to acquired mutations in some cases of early dissemination or may be microenvironmentally dictated in cases of late dissemination, where primary tumors and their metastases are genetically closely related [6,7,8]. The question regarding the temporal occurrence of metastasis is a controversial one, and most likely, different types of cancer display different progression trajectories to systemic disease with the microenvironment being a central player either via direct effects to DTCs or indirectly by providing the optimal conditions for acquisition of genetic changes.
This review aims to discuss the phenomenon of therapeutic pan-resistance of DTCs, micrometastases, and macrometastases. We will focus on the concept of cellular dormancy and its implications for resistance to chemotherapy and immunotherapy, as well as the role of the dynamic crosstalk of the tumor with the metastatic microenvironment (MME) and the immune system. Although significant uncertainties remain, a plethora of recent studies aided by novel experimental platforms have provided significant insight into the mechanisms that allow DTCs and micrometastases to resist chemotherapy and immunotherapy during dormancy and then outgrow into lethal macrometastases.
2. The Invasion-Metastasis Cascade
From an evolutionary perspective, metastasis can be thought of as a linear sequence of events, collectively described in the literature as the invasion-metastasis cascade [9]. In order for the cancer cells to arrive at the site of metastasis, they have to undergo a series of adaptations, including local invasion, intravasation, bloodborne dissemination, extravasation, and colonization, as well as coping with foreign environments much different from their tissue of origin [10]. At this point, cells that arrive in the metastatic setting exist either as DTCs or micrometastases [11]. While DTCs are solitary, dormant cells in a truly quiescent state, micrometastases most likely exist in a state of punctuated quiescence where proliferation is not continuous but rather sporadic before being suppressed, in contrast to previous assumptions in favor of a continuous balanced equilibrium between cell division and apoptosis [12]. It has to be emphasized that the aforementioned adaptations are based on stochastic events, and consequently, there is a high attrition rate of cells in hostile environments rendering metastasis an inefficient process [13,14]. Eventually, secondary to certain local or systemic events, DTCs or micrometastatic deposits exit from dormancy and start proliferating, giving rise to the actively growing, vascularized, lethal macrometastases [15].
In this context, dormancy and reawakening provide a solid explanation for the long periods of apparent stability seen in many cases of cancer, including breast cancer, prostate cancer, and melanoma [10]. Strikingly, excess mortality in breast cancer patients can be documented up to 20 years after surgery [16], while circulating breast cancer cells have been detected in patients clinically free of disease up to 22 years after diagnosis [17]. An interesting observation is that patients with HER2+ or triple negative (TN) breast cancer tend to relapse early, within five years from surgery, while ER+ cancers show a relatively stable rate of relapse over a period of several years [18,19]. In a similar fashion, in prostate cancer the median time from biochemical only recurrence (defined as an increase in prostate-specific antigen; PSA) after radical prostatectomy to bone metastasis and death exceed 16 years [20]. Such late and ultra-late recurrences are frequently seen in other neoplasms, such as melanoma and renal cell carcinoma [21,22], and illustrate the sequelae of the invasion-metastasis cascade as well as highlight the difficulty of efficiently treating DTCs and micrometastases.
3. Cancer-Associated Epithelial-Mesenchymal Plasticity
Epithelial-mesenchymal transition (EMT) is a biologic process with established roles in numerous developmental programs involved in new tissue and organ generation and is usually followed by the reverse process, mesenchymal-epithelial transition (MET) [23,24,25]. Cancer cells exploit these processes, namely cancer-associated epithelial-mesenchymal transition (cEMT) and cancer-associated mesenchymal-epithelial reverting transition (cMErT) to obtain a number of the classical hallmarks of cancer [26,27], mainly increased metastatic potential and enhancement of the cancer stem cell (CSC) phenotype [28,29,30]. It has to be emphasized that at any given time point cancer cells are not fixed either in a purely epithelial or mesenchymal state, but rather at some point along the epithelial-mesenchymal axis, harnessing beneficial properties from both polar states [31]. As a result, many scientists prefer the term cancer-associated epithelial-mesenchymal plasticity (cEMP) underpinning the hybrid state of these cells and the bidirectional potential based on internal and external cues.
The foundation for the role of cEMP in metastasis arises from observations and functional evidence of the increased ability of mesenchymally shifted carcinoma cells to escape from the primary tumor, along with acquisition of increased survival, stemness, and metastasis-initiating capacity compared to cancer cells with epithelial characteristics [23]. Nevertheless, the fact that induction of stable mesenchymal features abrogates metastatic outgrowth in preclinical models, together with clinical observations that metastases exhibit similar (or even enhanced) epithelial properties relative to their primary tumors, complicates the importance of cEMP in metastasis [28,32,33]. cEMP is also linked to stemness of tumor cells, which confers features supportive of successful outgrowth of metastatic deposits. Consistently, breast cancer cells that present a CSC phenotype frequently exhibit cEMT, and the abrogation of this process impinges on their stemness [34,35,36,37]. This relationship is not that straightforward though, as studies have shown an uncoupling of stemness from the mesenchymal state, and more specifically knockdown of the cEMT transducer PRRX1 reversed cEMT and promoted metastasis in breast cancer cells, with the carcinoma derived from this cMErT retaining stemness properties [38].
4. Role of E-Cadherin: Friend or Foe?
While cEMT is considered essential for successful tumor cell migration and invasion, it is now well accepted that cMErT confers increased survival and aids in successful colonization of metastatic sites by cancer cells [1]. Within this context, E-cadherin, a calcium-dependent cell-cell adhesion protein traditionally viewed as a tumor suppressor, seems to have a powerful role in therapeutic pan-resistance of micrometastases.
Early evidence for the importance of cMErT with regards to metastatic outgrowths came from observations of equal or increased levels of E-cadherin expression in distant metastases compared to their matched primary breast and prostate cancer specimens [39,40,41,42,43]. On top of that, metastatic tumors of invasive ductal adenocarcinoma origin express E-cadherin regardless of the E-cadherin status of the primary breast tumors [42]. Consistently, bone metastases from E-cadherin-negative, poorly differentiated breast carcinomas also exhibit E-cadherin expression [44]. All the above add one more layer of complexity regarding the role of cEMT in the cancer cell adaptation and establishment at the metastatic sites, instead pinpointing to E-cadherin as an essential player for the establishment and protection of metastatic deposits.
An abundance of markers have been associated with cEMP, including mesenchymal markers such as vimentin and N-cadherin, core cEMT-activating transcription factors (SNAIL1/2, ZEB1/2, TWIST1/2), and loss of epithelial protein expression (E-cadherin, EpCAM) (31). Of these, E-cadherin might be the strongest, since its loss is a hallmark of cEMT and strongly correlates with dissemination even in the absence of mesenchymal marker expression [27,45]. This is the basis for the partial epithelial and mesenchymal phenotypes noted in cancer cells.
The role of E-cadherin is extremely important in “shielding” micrometastases from chemotherapy as shown by Ma et al. in two studies in prostate cancer, where it was shown that hepatocytes induce a cMErT of prostate cancer cells via initial suppression of the MAP kinases p38 and ERK, conferring an epithelioid morphology to prostate cancer cells with an increase/re-expression of E-cadherin and other epithelial markers (ZO-1, connexin 43). Upon hepatocyte-dictated E-cadherin upregulation in prostate cancer cells and cell-cell adhesion through homeotypic binding, prostate cancer cells reactivated canonical survival pathways, such as PI3K/Akt, MEK/ERK, and JAK/STAT in response to chemotherapy and/or death signals, gaining resistance to treatment in a proliferation-independent manner [46,47] (Figure 1). The translational impact of these findings is impressive, as these pathways could be targeted in the context of combinatorial therapeutic approaches to synergistically improve the efficacy of traditional chemotherapies by using small doses of MEK inhibitors not for their direct therapeutic effect, but rather for re-sensitizing small micrometastases to existing chemotherapeutic agents.
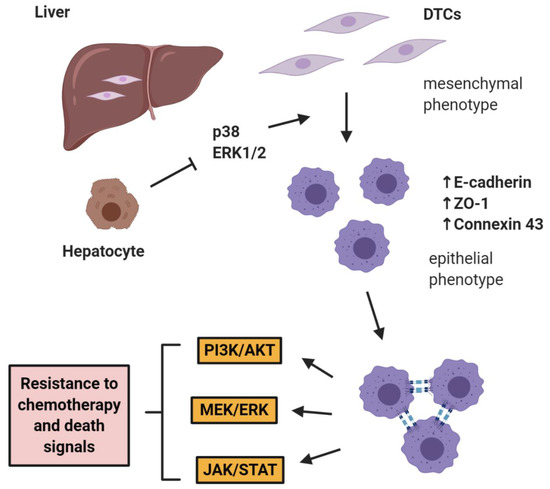
Figure 1. E-cadherin mediates micrometastatic dormancy and chemoresistance. Hepatocytes induce E-cadherin upregulation in metastatic prostate cancer as well as other epithelial markers, thus promoting a partial cancer-associated mesenchymal-to-epithelial reverting transition (cMErT) by initial suppression of p38 and ERK. Upon cell-cell E-cadherin ligandation, stimulation of metastatic cells with chemotherapy induces activation of canonical pro-survival kinases resulting in increased chemoresistance. This chemoresistance is proliferation-independent and can be targeted with an adjuvant treatment to achieve optimal efficacy with traditional chemotherapies. DTCs: disseminated tumor cells.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21197304