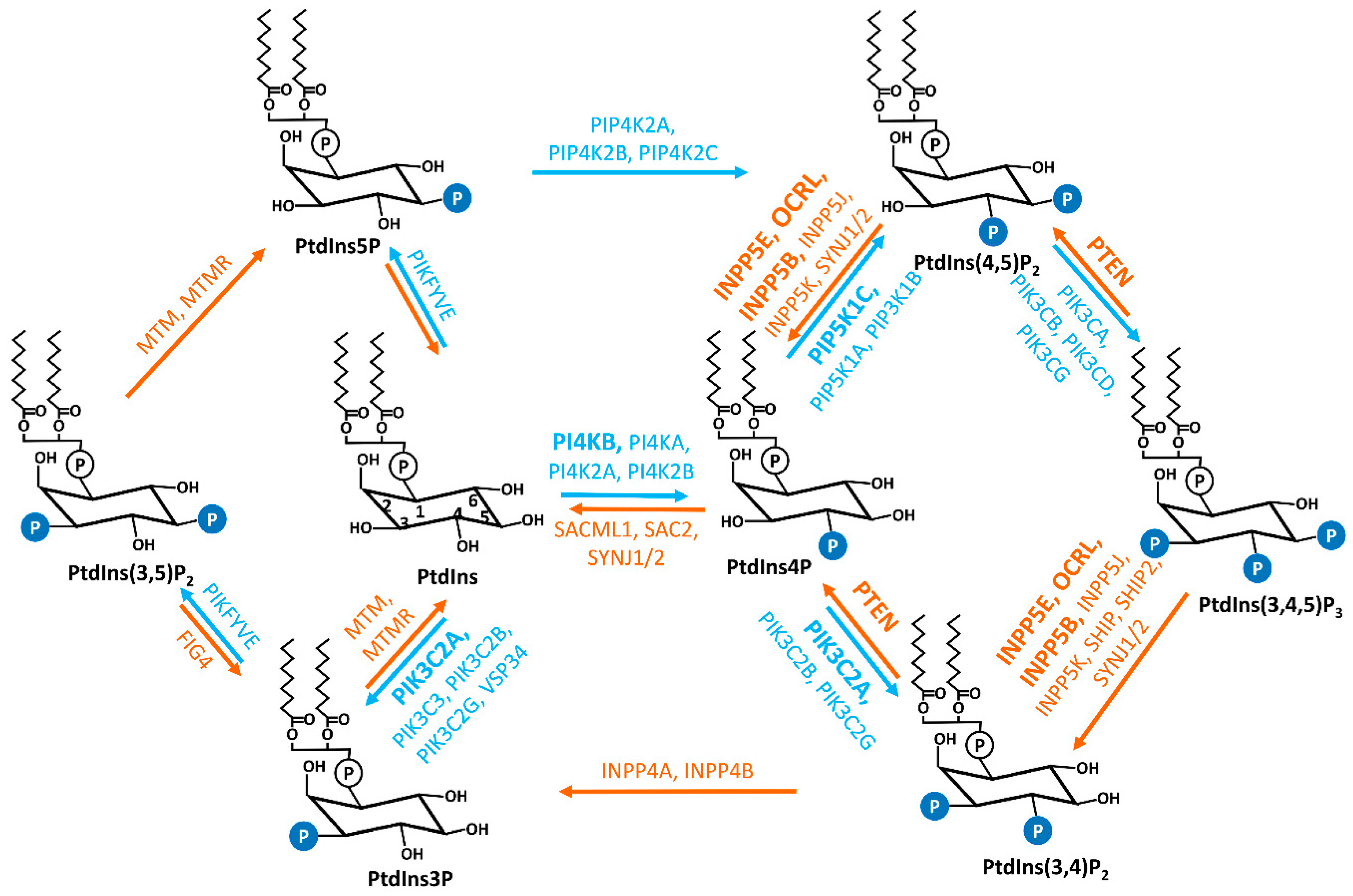
Primary cilia are microtube-based organelles that extend from the cell surface and function as biochemical and mechanical extracellular signal sensors. Primary cilia coordinate a series of signaling pathways during development. Cilia dysfunction leads to a pleiotropic group of developmental disorders, termed ciliopathy. Phosphoinositides (PIs), a group of signaling phospholipids, play a crucial role in development and tissue homeostasis by regulating membrane trafficking, cytoskeleton reorganization, and organelle identity. Accumulating evidence implicates the involvement of PI species in ciliary defects and ciliopathies. The abundance and localization of PIs in the cell are tightly regulated by the opposing actions of kinases and phosphatases, some of which are recently discovered in the context of primary cilia.
- phosphoinositide kinases and phosphatases
- primary cilia
1. Introduction
2. Primary Cilia
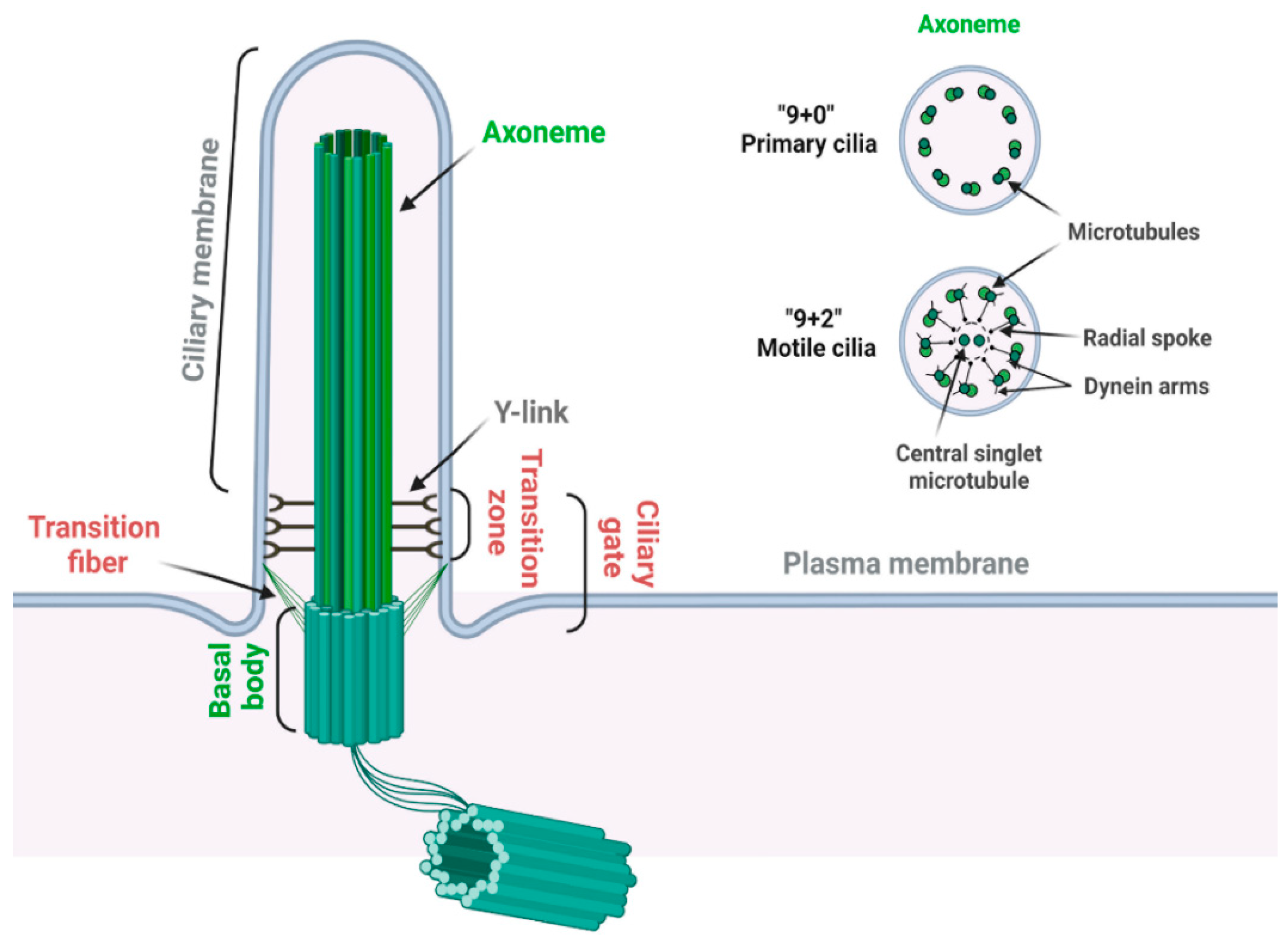
2.1. Structure of Primary Cilia
2.2. Ciliopathy
This entry is adapted from the peer-reviewed paper 10.3390/jdb10040051
References
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137.
- Schink, K.O.; Tan, K.W.; Stenmark, H. Phosphoinositides in Control of Membrane Dynamics. Annu. Rev. Cell Dev. Biol. 2016, 32, 143–171.
- Dickson, E.J.; Hille, B. Understanding phosphoinositides: Rare, dynamic, and essential membrane phospholipids. Biochem. J. 2019, 476, 1–23.
- De Craene, J.O.; Bertazzi, D.L.; Bar, S.; Friant, S. Phosphoinositides, Major Actors in Membrane Trafficking and Lipid Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 634.
- Posor, Y.; Jang, W.; Haucke, V. Phosphoinositides as membrane organizers. Nat. Rev. Mol. Cell Biol. 2022, 23, 797–816.
- Conduit, S.E.; Vanhaesebroeck, B. Phosphoinositide lipids in primary cilia biology. Biochem. J. 2020, 477, 3541–3565.
- Hammond, G.R.V.; Burke, J.E. Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr. Opin. Cell Biol. 2020, 63, 57–67.
- Kutateladze, T.G. Translation of the phosphoinositide code by PI effectors. Nat. Chem. Biol. 2010, 6, 507–513.
- Hammond, G.R.; Balla, T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta 2015, 1851, 746–758.
- Cullen, P.J.; Cozier, G.E.; Banting, G.; Mellor, H. Modular phosphoinositide-binding domains—Their role in signalling and membrane trafficking. Curr. Biol. 2001, 11, R882–R893.
- Lemmon, M.A. Phosphoinositide recognition domains. Traffic 2003, 4, 201–213.
- Rusten, T.E.; Stenmark, H. Analyzing phosphoinositides and their interacting proteins. Nat. Methods 2006, 3, 251–258.
- Jacobsen, R.G.; Mazloumi Gavgani, F.; Edson, A.J.; Goris, M.; Altankhuyag, A.; Lewis, A.E. Polyphosphoinositides in the nucleus: Roadmap of their effectors and mechanisms of interaction. Adv. Biol. Regul. 2019, 72, 7–21.
- Staiano, L.; De Leo, M.G.; Persico, M.; De Matteis, M.A. Mendelian disorders of PI metabolizing enzymes. Biochim. Biophys. Acta 2015, 1851, 867–881.
- Liu, Y.; Bankaitis, V.A. Phosphoinositide phosphatases in cell biology and disease. Prog. Lipid Res. 2010, 49, 201–217.
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156.
- Dickson, E.J. Recent advances in understanding phosphoinositide signaling in the nervous system. F1000Res 2019, 8, F1000 Faculty Rev-278.
- Krajnik, A.; Brazzo, J.A., 3rd; Vaidyanathan, K.; Das, T.; Redondo-Munoz, J.; Bae, Y. Phosphoinositide Signaling and Mechanotransduction in Cardiovascular Biology and Disease. Front. Cell Dev. Biol. 2020, 8, 595849.
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175.
- Plotnikova, O.V.; Pugacheva, E.N.; Golemis, E.A. Primary cilia and the cell cycle. Methods Cell Biol. 2009, 94, 137–160.
- Dutta, P.; Ray, K. Ciliary membrane, localised lipid modification and cilia function. J. Cell Physiol. 2022, 237, 2613–2631.
- Bielas, S.L.; Silhavy, J.L.; Brancati, F.; Kisseleva, M.V.; Al-Gazali, L.; Sztriha, L.; Bayoumi, R.A.; Zaki, M.S.; Abdel-Aleem, A.; Rosti, R.O.; et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 2009, 41, 1032–1036.
- Jacoby, M.; Cox, J.J.; Gayral, S.; Hampshire, D.J.; Ayub, M.; Blockmans, M.; Pernot, E.; Kisseleva, M.V.; Compere, P.; Schiffmann, S.N.; et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 2009, 41, 1027–1031.
- Dyson, J.M.; Conduit, S.E.; Feeney, S.J.; Hakim, S.; DiTommaso, T.; Fulcher, A.J.; Sriratana, A.; Ramm, G.; Horan, K.A.; Gurung, R.; et al. INPP5E regulates phosphoinositide-dependent cilia transition zone function. J. Cell Biol. 2017, 216, 247–263.
- Conduit, S.E.; Ramaswamy, V.; Remke, M.; Watkins, D.N.; Wainwright, B.J.; Taylor, M.D.; Mitchell, C.A.; Dyson, J.M. A compartmentalized phosphoinositide signaling axis at cilia is regulated by INPP5E to maintain cilia and promote Sonic Hedgehog medulloblastoma. Oncogene 2017, 36, 5969–5984.
- Xu, Q.; Zhang, Y.; Wei, Q.; Huang, Y.; Hu, J.; Ling, K. Phosphatidylinositol phosphate kinase PIPKIgamma and phosphatase INPP5E coordinate initiation of ciliogenesis. Nat. Commun. 2016, 7, 10777.
- Chavez, M.; Ena, S.; Van Sande, J.; de Kerchove d’Exaerde, A.; Schurmans, S.; Schiffmann, S.N. Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Dev. Cell 2015, 34, 338–350.
- Garcia-Gonzalo, F.R.; Phua, S.C.; Roberson, E.C.; Garcia, G., 3rd; Abedin, M.; Schurmans, S.; Inoue, T.; Reiter, J.F. Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Dev. Cell 2015, 34, 400–409.
- Zhang, R.; Tang, J.; Li, T.; Zhou, J.; Pan, W. INPP5E and Coordination of Signaling Networks in Cilia. Front. Mol. BioSci. 2022, 9, 885592.
- Nakatsu, F. A Phosphoinositide Code for Primary Cilia. Dev. Cell 2015, 34, 379–380.
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344.
- Linck, R.W.; Chemes, H.; Albertini, D.F. The axoneme: The propulsive engine of spermatozoa and cilia and associated ciliopathies leading to infertility. J. Assist. Reprod. Genet. 2016, 33, 141–156.
- Satir, P. CILIA: Before and after. Cilia 2017, 6, 1.
- Goncalves, J.; Pelletier, L. The Ciliary Transition Zone: Finding the Pieces and Assembling the Gate. Mol. Cells 2017, 40, 243–253.
- Wei, Q.; Ling, K.; Hu, J. The essential roles of transition fibers in the context of cilia. Curr. Opin. Cell Biol. 2015, 35, 98–105.
- Garcia-Gonzalo, F.R.; Reiter, J.F. Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Csh Perspect. Biol. 2017, 9, a028134.
- Garcia-Gonzalo, F.R.; Reiter, J.F. Scoring a backstage pass: Mechanisms of ciliogenesis and ciliary access. J. Cell Biol. 2012, 197, 697–709.
- Nachury, M.V. How do cilia organize signalling cascades? Philos. Trans. R. Soc. Lond B Biol. Sci. 2014, 369, 20130465.
- Lee, K.H. Involvement of Wnt signaling in primary cilia assembly and disassembly. Febs. J. 2020, 287, 5027–5038.
- Wallingford, J.B. Planar cell polarity signaling, cilia and polarized ciliary beating. Curr. Opin. Cell Biol. 2010, 22, 597–604.
- Schou, K.B.; Pedersen, L.B.; Christensen, S.T. Ins and outs of GPCR signaling in primary cilia. Embo. Rep. 2015, 16, 1099–1113.
- Christensen, S.T.; Clement, C.A.; Satir, P.; Pedersen, L.B. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J. Pathol. 2012, 226, 172–184.
- Sreekumar, V.; Norris, D.P. Cilia and development. Curr. Opin. Genet. Dev. 2019, 56, 15–21.
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219.
- Ferkol, T.W.; Leigh, M.W. Ciliopathies: The central role of cilia in a spectrum of pediatric disorders. J. Pediatr. 2012, 160, 366–371.
- Quinlan, R.J.; Tobin, J.L.; Beales, P.L. Modeling ciliopathies: Primary cilia in development and disease. Curr. Top Dev. Biol. 2008, 84, 249–310.
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893.
- Focsa, I.O.; Budisteanu, M.; Balgradean, M. Clinical and genetic heterogeneity of primary ciliopathies (Review). Int. J. Mol. Med. 2021, 48, 176.
- Powles-Glover, N. Cilia and ciliopathies: Classic examples linking phenotype and genotype-an overview. Reprod. Toxicol. 2014, 48, 98–105.
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Csh Perspect. Biol. 2017, 9, a028191.
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547.
- Wheway, G.; Mitchison, H.M.; Genomics England Research, C. Opportunities and Challenges for Molecular Understanding of Ciliopathies-The 100,000 Genomes Project. Front. Genet. 2019, 10, 127.