Primary cilia are microtube-based organelles that extend from the cell surface and function as biochemical and mechanical extracellular signal sensors. Primary cilia coordinate a series of signaling pathways during development. Cilia dysfunction leads to a pleiotropic group of developmental disorders, termed ciliopathy. Phosphoinositides (PIs), a group of signaling phospholipids, play a crucial role in development and tissue homeostasis by regulating membrane trafficking, cytoskeleton reorganization, and organelle identity. Accumulating evidence implicates the involvement of PI species in ciliary defects and ciliopathies. The abundance and localization of PIs in the cell are tightly regulated by the opposing actions of kinases and phosphatases, some of which are recently discovered in the context of primary cilia.
1. Introduction
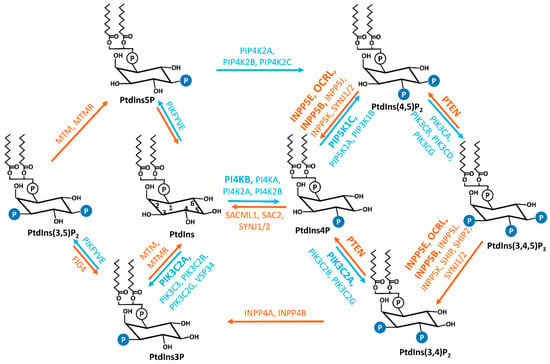
Phosphoinositides (PIs) are reversibly phosphorylated derivatives of the membrane phosphatidylinositol (PtdIns,
Figure 1) [
1,
2]. PIs consist of two fatty acid chains, a glycerol moiety, and a D-myo-inositol-1-phosphate head group that is decorated by phosphate moiety at the 3-, 4-, and/or 5-hydroxyl position to produce seven phosphorylated derivatives (PtdIns3P, PtdIns4P, PtdIns5P, PtdIns(3,4)P
2, PtdIns(3,5)P
2, PtdIns(4,5)P
2, and PtdIns(3,4,5)P
3) [
1]. The exact composition of each species varies in different subcellular locations and different cell types. Compared to other phospholipids in cellular membrane compartments, PIs are relatively minor constituents and PtdIns4P and PtdIns(4,5)P
2, which contribute the largest amounts, are less than 1% of the total cellular phospholipid pool [
1]. Despite the low abundance, PIs are indispensable to the integrity of cells. PIs play essential roles in various cellular events including, but not limited to, cytoskeletal dynamics, cell adhesion and migration, vesicular trafficking, assembly of cargo proteins at membranes, cell proliferation, and survival [
1,
2,
3,
4]. PIs are key identity determinants of various cellular membrane compartments and act as spatiotemporal cues to direct signaling events [
2,
3,
5,
6,
7].
Figure 1. Interconversions of seven PIs. The reversible production of the PIs is mediated by the combined activities of specific phosphatases and kinases, which are labeled in orange and blue, respectively. The kinases and phosphatases that have been reported to be associated with cilia are marked in bold. PtdIns, phosphatidylinositol; PtdIns3P, phosphatidylinositol 3-phosphate; PtdIns4P, phosphatidylinositol 4-phosphate; PtdIns5P, phosphatidylinositol 5-phosphate; PtdIns(3,4)P2, phosphatidylinositol 3,4-phosphate; PtdIns(3,5)P2, phosphatidylinositol 3,5-phosphate; PtdIns(4,5)P2, phosphatidylinositol 4,5-phosphate; PtdIns(3,4,5)P3, phosphatidylinositol 3,4,5-trisphosphate; PI4KA, phosphatidylinositol 4-kinase alpha; PI4KB, phosphatidylinositol 4-kinase beta; PI4K2A, phosphatidylinositol 4-kinase type 2 alpha; PI4K2B, phosphatidylinositol 4-kinase type 2 beta; PIK3C3, phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3C2A, phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 alpha; PIK3C2B, phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 beta; PIK3C2G, phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 gamma; PIP5K1A, phosphatidylinositol-4-phosphate 5-kinase type 1 alpha; PIP5K1B, phosphatidylinositol-4-phosphate 5-kinase type 1 beta; PIP5K1C, phosphatidylinositol-4-phosphate 5-kinase type 1 gamma; PIP4K2A, phosphatidylinositol-5-phosphate 4-kinase type 2 alpha; PIP4K2B, phosphatidylinositol-5-phosphate 4-kinase type 2 beta; PIP4K2C, phosphatidylinositol-5-phosphate 4-kinase type 2 gamma; PIKFYVE, phosphoinositide kinase, FYVE-type zinc finger containing; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PIK3CB, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta; PIK3CD, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta; PIK3CG, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma; MTM, myotubularin; MTMR, myotubularin-related; SAC2, Sac domain-containing inositol phosphatase 2; SACML1, SAC1 like phosphatidylinositide phosphatase; SYNJ1/2, synaptojanin 1/2; INPP5E, inositol polyphosphate-5-phosphatase E; INPP5J, inositol polyphosphate-5-phosphatase J; OCRL, oculocerebrorenal syndrome of Lowe; INPP5B, inositol polyphosphate-5-phosphatase B; INPP5K, inositol polyphosphate-5-phosphatase K; PTEN, phosphatase and tensin homolog; INPP4A, inositol polyphosphate-4-phosphatase type I A; INPP4B, inositol polyphosphate 4-phosphatase type II; FIG4, factor-induced gene 4; SHIP, Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase.
Each PI species exhibits unique distribution across the different plasma membrane (PM) and organelle membrane compartments [
2]. In general, the lipid tail of PIs inserts into membrane compartments, whereas the phosphorylated inositol head of individual PIs recognizes and binds to unique PI effector proteins [
2,
8,
9]. Current reported PI-binding domains include the Pleckstrin Homology (PH), FYVE, Phox (PX), C2, PROPPIN, PTB, Tubby, TRAF, ANTH/ENTH, FERM, and PDZ domains [
9,
10,
11]. The affinity and specificity vary between different PIs and binding domains [
9]. Individual PI species induce the conformational change of effectors and/or recruits them to specific subcellular locales to meet their signaling partners [
8,
9,
12,
13]. This leads to the activation/inhibition of PI effectors at unique subcellular locales [
8]. Thus, the precisely controlled spatiotemporal availability of PI species in the cell is critical. PIs metabolism and homeostasis are maintained and regulated by specific PI kinases and phosphatases that are conserved in evolution [
3,
5,
8]. Many reports have discovered that the dysfunction of components of the PI signaling pathway is responsible for various human diseases, from rare genetic disorders to the most common conditions such as cancer, obesity, neurological disorders, cardiovascular disease, and diabetes [
7,
14,
15,
16,
17,
18].
The primary cilium is a sensory organelle presenting on the surface of most mammalian cell types [
19]. Although the ciliary membrane is reported to be continuous with the plasma membrane, the lipid and protein composition in the ciliary membrane is maintained unique and distinguishable from the plasma membrane [
20,
21]. To fulfill its function as a signaling hub and cellular antenna, the trafficking of protein and membrane components in and out of cilia is highly active and tightly controlled, suggesting the potential involvement of PIs in cilia. Indeed, it was reported in 2009 that mutations in an inositol polyphosphate 5-phosphatase, INPP5E, are causal for multiple ciliopathies [
22,
23]. Subsequently, more studies implicated the connection between PIs and primary cilia. PIs are reported to regulate the stability/disassembly of primary cilia [
24,
25], ciliogenesis [
26], and ciliary trafficking [
27,
28]. The dysfunction of multiple PI enzymes, both phosphatases, and kinases, is connected to ciliary defects and ciliopathies [
6]. The localization and function of PIs in cilia have been reviewed in several excellent recent reviews [
6,
29,
30].
2. Primary Cilia
2.1. Structure of Primary Cilia
The primary cilium is a sensory organelle presenting on the surface of most quiescent cells in vertebrates [
19,
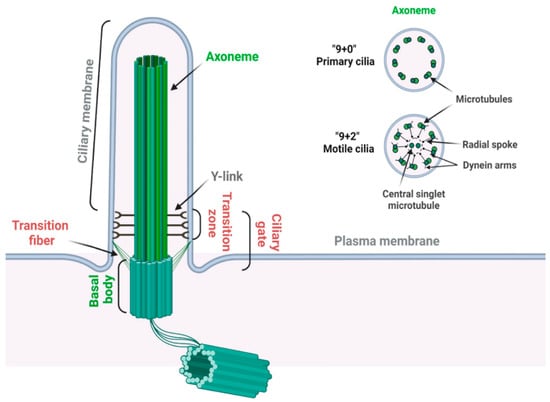
31]. The primary cilium is composed of a microtubule-based core structure called the axoneme, which is surrounded by a ciliary membrane that is continuously extended from the plasma membrane (
Figure 2) [
32]. Different from motile cilia, which can generate a fluid flow on cells such as respiratory epithelial and ependymal cells, primary cilia only contain nine microtubule doublets without two centrally located singlet microtubules (
Figure 2) and are non-motile for lacking the axonemal dynein arms on the outer microtubules [
33].
Figure 2. Structure of the primary cilium. A primary cilium is composed of a “9 + 0” microtubule axoneme and basal body complex (labeled in green). A motile cilium has two extra central singlet microtubules, generating a “9 + 2” arrangement (labeled in green). A motile cilium also contains dynein arms for ciliary movement and radial spoke to regulate the motility and motion pattern of motile cilia (labeled in black). Transition zone is characterized by the Y-links, which connect the proximal axoneme to the ciliary membrane. Transition fibers connect the distal end of the basal body to the ciliary membrane. Transition zone and transition fibers together coordinate the ciliary gate function (labeled in red) (created with
BioRender.com, accessed on 18 November 2022).
The basal body, transformed from the mother centriole upon serum starvation, nucleates the outgrowth of the axoneme and forms the base of primary cilia. The region between the axoneme and basal body is identified as the transition zone, which is characterized by Y-links connecting the axonemal microtubules to the ciliary membrane [
34]. Transition fibers, which are transformed from distal appendages of the mother centriole, are located below the transition zone on the distal end of the basal body [
35]. In addition to anchoring the basal body to the plasma membrane, transition fibers function together with the transition zone as the ciliary gate to regulate ciliogenesis and signaling by controlling the selective transport of specific ciliary proteins between the cell body and the cilium [
35,
36]. Intriguingly, many transition zone and transition fiber proteins are reported to contain phosphoinositide binding domains; however, the physiological significance of their PI binding potential remains unclear [
37].
2.2. Ciliopathy
Although largely considered a vestige before 1999, the primary cilium is now demonstrated as a signaling center to interpret both chemical and mechanical signals. Major components of multiple signaling pathways specifically localize in primary cilia [
38]. These include pathways that are essential for the biogenesis and homeostasis of tissues and organs, such as the Hedgehog signaling [
19], canonical Wnt signaling [
39], Planar cell polarity [
40], G protein-coupled receptor signaling [
41], as well as tyrosine kinase receptor signaling [
42] pathways. The activation and regulation of these pathways occur in cilia and depend on the normal structure and functionality of primary cilia [
19].
Consistently, defects in primary cilia lead to a panel of genetic human diseases, collectively termed ciliopathies, including numerous seemingly unrelated syndromes, with involvement of the brain, eye, kidney, liver, pancreas, skeletal system, and some others in a growing list [
43,
44,
45,
46,
47,
48]. Common ciliopathies range from organ-specific disorders such as autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive PKD (ARPKD) to pleiotropic disorders such as Joubert syndrome (JBST), nephronophthisis (NPHP), Bardet–Biedl syndrome (BBS), and Meckel syndrome (MKS) [
49,
50]. Currently, more than 190 established and 250 candidate cilium-associated proteins are identified as ciliopathy proteins, and over 35 ciliopathies are reported, emphasizing the significance of primary cilia in development and human disease [
51,
52]. Given the physiological significance of the primary cilium, the molecular mechanisms underlying its biogenesis, function, and related disease pathogenesis have just started to be explored in the recent two decades.
This entry is adapted from the peer-reviewed paper 10.3390/jdb10040051