Diabetic cardiomyopathy (DCM), a diabetes-induced microvascular complication, is defined as a heart disease in diabetes patients, which results in a structurally and functionally abnormal myocardium in the absence of hypertension, coronary artery disease, and congenital or valvular heart disorders [
1]. Approximately 12% of diabetes patients have DCM, which is the main cause of death [
2,
3]. The main clinical features of DCM are myocardial remodeling, diastolic and systolic dysfunction, and poor prognosis for diabetes patients, which can ultimately result in clinical heart failure (HF) [
2,
4]. HF can occur in both type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM), and DCM accounts for 44% of deaths in T1DM patients and 52% of deaths in T2DM patients [
5]. DCM development and progression are associated with increased myocardial metabolic abnormalities, myocardial apoptosis, autophagy, pyroptosis and ferroptosis, oxidative stress (OS), inflammation, cardiac fibrosis, and microangiopathy [
1,
6,
7].
2. Role of lncRNAs in Various Types of Cardiomyocyte Death in DCM
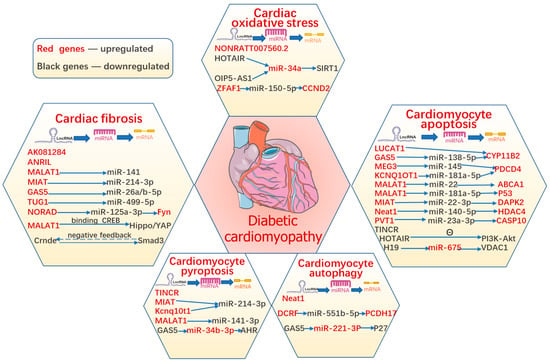
Evidence suggests that various types of cardiomyocyte death simultaneously act as terminal pathways in DCM and eventually accelerate the development of structural and functional impairment in HF, including apoptosis, autophagic cell death, pyroptosis, ferroptosis, and necroptosis. Until now, the suppression of any form of cardiomyocyte death has had a protective function in DCM; however, the regulatory mechanisms of cell death remain unclear and require further clarification. In addition, lncRNAs have been reported to participate in diabetes-induced cardiomyocyte death (Figure 3).
Figure 3. The role of lncRNAs in diabetic cardiomyopathy.
LncRNAs regulate various pathophysiological processes in diabetic cardiomyopathy, including cardiomyocyte apoptosis, pyroptosis, autophagy, and ferroptosis, as well as cardiac oxidative stress and fibrosis. The summary figure shows information on the stud-ies about lncRNAs in DCM.
3. Role of lncRNAs in Oxidative Stress in DCM
Hyperglycemia can induce excess ROS generation through AGEs, the polyol pathway, and de novo synthesis of triose metabolism [
107]. Significantly increased ROS levels are a pathological feature of DCM [
108] and can overwhelm their removal mechanisms, leading to OS [
7]. In DCM, the unbalanced redox state aggravates irreversible damage and death of cardiomyocytes, ultimately leading to cardiac dysfunction [
109]. Studies have confirmed that antioxidant interventions to scavenge ROS inhibit or prevent cardiac dysfunction in diabetic animal models [
110,
111,
112]. Recent evidence suggests that targeting OS using lncRNAs may be a promising approach for DCM. Yu et al. detected the expression of lncRNAs in HG-treated cardiomyocytes and identified the lncRNA NONRATT007560.2 as one of the top three upregulated lncRNAs [
113]. Furthermore, inhibition of NONRATT007560.2 lowered the generation of ROS in HG-treated cardiomyocytes, suggesting NONRATT007560.2 can inhibit diabetes-induced myocardial OS.
Sirtuin 1 (SIRT1) is a redox-sensitive enzyme that appears to improve DCM by targeting cellular factors and increasing stress resistance [
114]. SIRT1 upregulation attenuated ER stress-induced cardiomyocyte apoptosis [
115]. The lncRNAs OIP5-AS1 (Opa-interacting protein 5-antisense transcript 1) and HOTAIR were both significantly decreased in DCM [
116,
117]. Overexpression of HOTAIR or OIP5-AS1 improves cardiomyocyte viability and alleviates OS through the miR-34a/SIRT1 axis; therefore, they may be new therapeutic targets for DCM.
In 2012, Dixon [
118] first proposed a new concept of regulated cell death called ferroptosis, which is caused by the intracellular deposition of iron and lipid peroxide after excessive accumulation of ROS. In DCM, the essential factor in ferroptosis is OS injury [
118]. In vivo, the activation of nuclear factor erythroid 2-related factor 2 (NRF2) by sulforaphane (SFN) alleviated the progression of DCM by inhibiting myocardial cell ferroptosis [
119]. NRF2 plays a critical role in regulating the cellular antioxidant response by controlling the expression of many genes that counteract the effects of OS. The lncRNA zinc finger antisense 1 (ZFAS1) was shown to promote cardiomyocyte ferroptosis in DCM by sponging miR-150-5p and downregulating cyclin D2 (CCND2), which contributes to myocardial repair by regulating the cell cycle [
120]. The regulation of lncRNAs during OS in DCM is summarized in
Table 3. The roles of lncRNAs in OS in DCM are summarized in
Figure 3.
4. Role of lncRNAs in Diabetes-Induced Cardiac Fibrosis
Cardiac fibrosis is an important cause of cardiac dysfunction in DCM patients. Abnormally elevated ECM deposition, in particular collagen, increases myocardial stiffness and results in ventricular remodeling and dysfunction of LV relaxation and contraction [
88]. Hyperglycemia activates the matrix-synthesis program in cardiac fibroblasts by the stimulation of transforming growth factor β (TGF-β) cascades, resulting in the accumulation of AGEs and AGE-mediated fibroblasts [
121,
122,
123]. Rapid activation of TGF-β exerts a broad range of direct effects on cardiac fibroblasts (CFs) [
124], including the activation of downstream Smad-dependent signaling cascades, the induction of myofibroblast conversion, and the accumulation of ECM [
125,
126]. Melatonin, a hormone produced by the pineal gland, has an anti-fibrotic effect on DCM pathogenesis. Melatonin can inhibit TGF-β1/Smad2/3 signaling and NLRP3 inflammasome activation, which can be mediated via the MALAT1/miR-141 axis [
127]. The lncRNA colorectal neoplasia differentially expressed (Crnde) is a cardiac-specific and CF-enriched lncRNA that has been found to be negatively correlated with the cardiac fibrosis marker genes in 376 human heart tissues [
128]. Overexpression of Crnde attenuated myofibroblast differentiation and cardiac fibrosis in DCM mice by inhibiting the transcriptional activation of Smad3. Interestingly, Smad3 also transcriptionally activated Crnde expression, indicating the existence of a delicate Smad3-Crnde negative feedback.
Inflammatory cytokines directly stimulate the recruitment and activation of lymphocytes and macrophages and promote the pathogenesis of cardiac fibrosis. Myriad proinflammatory cytokines and chemokines are secreted by proinflammatory macrophages in injured cardiac tissues [
129]. Their crosstalk with fibroblasts promotes fibroblast differentiation into myofibroblasts, exacerbating extracellular matrix deposition [
130]. TGF-β stimulates NLRP3 expression and activates α-SMA, thereby promoting myofibroblast differentiation [
131]. IL-17 is a proinflammatory cytokine secreted by activated CD4+ T cells [
132]. IL-17 accelerates the production of IL-6 in cardiac fibroblasts, leading to myofibroblasts [
133]. The expression of IL-17 and lncRNA MIAT is significantly upregulated in the serum of diabetes patients [
132]. MIAT inhibits IL-17 production by specifically attenuating miR-214-3p in primary cardiac fibroblasts. Consequently, decreased IL-17 expression alleviates the onset of cardiac fibrosis and improves cardiac contractility. Ablation of IL-17 improved cardiac function and alleviated cardiac interstitial fibrosis by inhibiting the lncRNA AK081284 in diabetic mice [
134].
Vascular endothelial growth factor (VEGF) is activated by sustained metabolic and hemodynamic perturbations in diabetes. VEGF mediates ECM deposition and aggravates cardiac fibrosis by upregulating the pro-fibrotic growth factors TGFβ1 and connective tissue growth factor (CTGF) [
135]. The lncRNA ANRIL (antisense non-coding RNA in the INK4 locus) is a recruiter of the polycomb repressive complex (PRC) that facilitates the alteration of chromatin structure [
136]. Thomas et al. discovered that ANRIL promotes the synthesis of ECM and VEGF via epigenetic upregulation of EZH2 and the histone acetylator p300, deteriorating cardiac fibrosis in diabetic hearts [
137].
The Hippo pathway can negatively regulate the transcriptional coactivators Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), through the activation and phosphorylation of large tumor suppressor (LATS)1/2 kinases [
138]. Phosphorylation of LATS1 promotes YAP/TAZ nuclear export and abrogates transcriptional effects in the nucleus [
124]. YAP/TAZ is implicated in fibrotic actions by driving fibrosis-related target gene expression in the nucleus, accentuating TGF-β-driven activation of Smad2/3, and stimulating fibroblast proliferation [
124,
139]. Emerging evidence suggests that YAP/TAZ also affects cardiac fibrosis [
140,
141]. In HG CFs, MALAT1 and YAP expression in the nucleus was markedly increased and the phosphorylation of LATS1 was decreased [
142]. MALAT1 positively regulates YAP by binding to cAMP-responsive element-binding protein (CREB). Furthermore, Liu et al. confirmed that MALAT1 knockdown alleviated collagen accumulation and diabetic fibrosis through the Hippo pathway/YAP signaling pathway [
142].
The lncRNAs, TUG1 (taurine upregulated gene 1), NORAD (non-coding RNA activated by DNA damage), and GAS5 are upregulated in the myocardial tissues of diabetic mice [
143,
144,
145]. These lncRNAs exacerbate cardiac fibrosis by negatively regulating miR-499-5p, miR-125a-3p, and miR-26a/b-5p [
143,
144,
145].
Table 3 summarizes the lncRNAs implicated in the pathogenesis of cardiac fibrosis in DCM.
Figure 3 summarizes the vital roles of lncRNAs in cardiac fibrosis of DCM.
Table 3. LncRNAs implicated in the pathogenesis of oxidative stress and cardiac fibrosis in DCM.
| LncRNAs |
Experimental Model |
Target Genes |
Expression |
Mechanism Involved |
References |
| Oxidative Stress |
| NONRATT007560.2 |
HG-treated primary culture of neonatal cardiomyocytes |
|
upregulated |
inhibition of NONRATT007560.2 abated the formation of ROS |
[113] |
| HOTAIR |
STZ-induced diabetic rat model and HG-treated H9c2 cells |
miR-34a |
downregulated |
HOTAIR protected against
DCM via activation of the SIRT1 expression by sponging miR-34a |
[116] |
| OIP5-AS1 |
HG-treated H9c2 cells |
miR-34a |
downregulated |
OIP5-AS1 overexpression promoted viability and inhibits high glucose-induced oxidative stress of cardiomyocytes by targeting miRNA-34a/SIRT1 Axis |
[117] |
| ZFAS1 |
STZ-induced diabetic mouse model and primary culture of neonatal cardiomyocytes |
miR-150-5p |
upregulated |
inhibition of ZFAS1 attenuated ferroptosis by sponging miR-150-5p and activating CCND2 against DCM |
[120] |
| Cardiac Fibrosis |
| ZFAS1 |
STZ-induced diabetic mouse model and primary culture of neonatal cardiomyocytes |
miR-150-5p |
upregulated |
inhibition of ZFAS1 attenuated ferroptosis by sponging miR-150-5p and activating CCND2 against DCM |
|
| MALAT1 |
STZ-induced diabetic mice model and HG-treated primary culture of neonatal CFs |
miR-141 |
upregulated |
melatonin alleviated cardiac fibrosis via inhibiting MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-β1/Smads signaling |
[127] |
| Crnde |
human myocardial biopsies, STZ-induced diabetic mice model, and HG-treated primary culture of neonatal CFs |
|
downregulated |
lncRNA Crnde attenuated cardiac fibrosis via Smad3-Crnde negative feedback |
[128] |
| MIAT |
Human serum samples, STZ-induced diabetic mice model, and HG-treated primary culture of neonatal CFs |
miR-214-3p |
upregulated |
MIAT inhibited IL-17 production and alleviated the onset of cardiac fibrosis via specific attenuating miR-214-3p |
[132] |
| AK081284 |
STZ-induced diabetic mice model and HG-treated primary culture of neonatal CFs |
|
upregulated |
AK081284 knockdown inhibited the production of collagen I, collagen III, TGFβ1 and α-SMA stimulated by IL-17 |
[134] |
| ANRIL |
STZ-induced diabetic mice model |
|
upregulated |
ANRIL upregulated production of ECM proteins and VEGF via epigenetic upregulating p300 and EZH2 |
[137] |
| MALAT1 |
STZ-induced diabetic mice model and neonatal mouse HG-treated CFs |
CREB |
upregulated |
MALAT1 regulated diabetic cardiac fibroblasts through the Hippo/YAP signaling pathway by binding CREB |
[142] |
| TUG1 |
STZ-induced diabetic mice model and HG-treated cardiomyocytes |
miR-499-5p |
upregulated |
inhibition of TUG1 protected against DCM-induced diastolic dysfunction by regulating miR-499-5p |
[143] |
| NORAD |
subcutaneous injection of angiotensin II (ATII) in db/db mice and HG-treated primary mouse cardiomyocytes |
miR-125a-3p/Fyn |
upregulated |
silencing NORAD mitigated fibrosis and inflammatory responses via the ceRNA network of NORAD/miR-125a-3p/Fyn |
[144] |
| GAS5 |
STZ-induced diabetic mice model and HG-treated primary culture of neonatal cardiomyocytes |
miR-26a/b-5p |
upregulated |
silencing GAS5 alleviated apoptosis and fibrosis by targeting miR-26a/b-5p |
[145] |