Fatty acid metabolism provides energy for cell metabolism and growth by degrading fatty acids. Dysregulated fatty acid metabolism drives lipid synthesis and deposition and is associated with several cancers [
1]. Fatty acid metabolism is regulated by various enzymes [
2]. The long-chain fatty acyl CoA synthetase family (ACSLs) contains five isoforms identified as ACSL1, 3, 4, 5, and 6. They convert free long-chain fatty acids into fatty acyl-CoA esters and play a dominant role in both anabolic (fatty acid synthesis and lipogenesis) and catabolic pathways (lipolysis and fatty acid β-oxidation), although they have distinct substrate specificity, subcellular localization, and tissue distribution [
3]. ACSLs affect the behavior of malignant cells, including proliferation, migration, invasion, apoptosis, and drug resistance.
2. ACSL3 and ACSL4
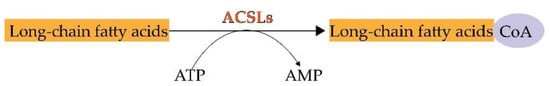
During lipid metabolism, the activation of fatty acids by esterification with coenzyme A is indispensable. ACSLs catalyze this reaction which is the first step in the utilization of fatty acids in mammals [
7] (
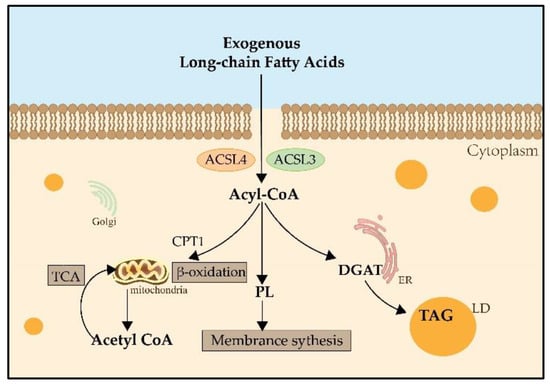
Figure 1). The main process of catalysis and the metabolic pathways of long-chain fatty acids are described in
Figure 2 [
8,
9]. Researchers have already found that ACSL3 and ACSL4 are involved in ferroptosis [
10]. However, information is limited regarding the specific roles of ACSL3 and 4 in ferroptosis.
Figure 1. The chemical reaction of ACSLs.
Figure 2. The metabolic pathways of long-chain fatty acids. Both ACSL3 and ACSL4 catalyze exogenous long-chain fatty acids into Acyl-CoA. Acyl-CoA can enter the mitochondria by the transportation of CPT1, then catalyze the formation of Acetyl-CoA and participate in the TCA for providing energy. Acyl-CoA also can produce PLs, which contribute to membrane synthesis. And Acyl-CoA can catalyze DGAT and be involved in the synthesis of TAG in the LDs for lipid storage. CPT1: carnitine palmitoyltransferase 1; TCA: tricarboxylic acid cycle; PLs: phospholipids; DGAT: Diglyceride Acyltransferase; TAG: triglyceride; LD: lipid droplet.
ACSL3 is highly expressed in the brain, testes, and skeletal muscle, and preferentially utilizes myristate, arachidonate, and eicosapentaenoate as substrates. ACSL3 mainly resides in the Golgi apparatus, endoplasmic reticulum (ER), peroxisomes, and mitochondria [
11]. ACSL3 is a lipid droplet (LD)-associated protein. Most cells have a large amount of ACSL3 on the surface of LDs, and this protein has a number of pathophysiological functions in LD formation, autophagy control, and cellular ferroptosis [
12]. It participates in several pathological processes, including LD production, autophagy control, and cellular ferroptosis.
ACSL4 is mainly located in the ER, mitochondria, plasma membrane, and peroxisome, and is enriched in the adrenal gland, ovary, testis, and brain tissues [
3]. Microarray analysis using a genome-wide CRISPR-based genetic screening system and iron death-resistant cell lines revealed that ACSL4 is required for the induction of ferroptosis through the accumulation of oxidized cell membrane phospholipids (PLs) [
5,
13].
3. ACSL3 and ACSL4 in Ferroptosis
Ferroptosis is characterized by excessive accumulation and failure to eliminate iron-dependent lethal toxic lipid ROS, thereby initiating cell death. As a type of RCD, ferroptosis is either directed by intracellular signals or induced by external factors, leading to the initiation of suicidal protective strategies. It is considered a highly complicated and strictly orchestrated process that is regulated by a series of signals from distinct cell organelles, such as mitochondria, ER, and lysosomes [
14].
Morphologically, distinct from other types of RCD such as apoptosis, necroptosis, and pyroptosis, ferroptosis shows swelling of cells, formation of pores in the cell membrane, smaller mitochondria, reduced mitochondrial cristae, increased mitochondrial membrane density, normal-sized nuclei and non-cohesive chromatin [
15].
Ferroptosis is initiated by the accumulation of iron, lipid peroxidation, and subsequent plasma membrane rupture. Lipid peroxidation is a free radical-driven reaction that primarily affects polyunsaturated fatty acids (PUFA) in the cell membrane and undergoes three stages including initiation, propagation, and termination. Finally, lipid peroxidation produces lipid hydroperoxides [
14]. Iron can favor tumor incidence and growth, as it is an important nutrient for cell proliferation and a key cofactor of metabolic enzymes [
16]. Cellular or organelle membranes are particularly susceptible to lipid peroxidation because of their high PUFA content [
17]. Cystatin-GSH-GPX4, CoQ10-FSP1, and GCH1-BH4-DHFR can effectively resist ferroptosis by inhibiting lipid peroxidation [
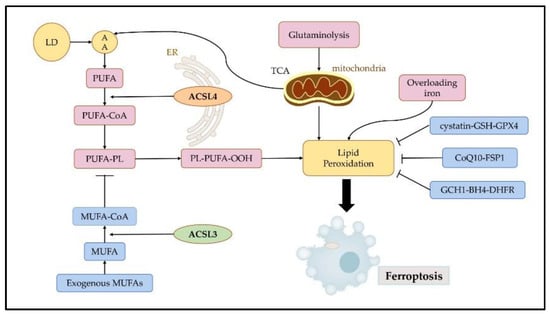
18] (
Figure 3).
Figure 3. Regulatory mechanisms of ferroptosis. Ferroptosis is mainly initiated by excessive production and failure to eliminate iron, lipid peroxidation, and subsequent plasma membrane rupture. Ferroptosis is induced by lipid peroxidation. ACSL4 contributes to the synthesis of high levels of PUFAs, which promote ferroptosis. ACSL3 is key to MUFA-induced ferroptosis resistance. Three pathways, namely the cystatin-GSH-GPX4, CoQ10-FSP1, and GCH1-BH4-DHFR axis can effectively resist ferroptosis by inhibiting lipid peroxidation. LDs: lipid droplets; AA: arachidonic acid; PUFA: polyunsaturated fatty acids; MUFA: monounsaturated fatty acids; ER: endoplasmic reticulum; TCA: tricarboxylic acid cycle.
Fatty acids belong to three categories: saturated fatty acids (no double bond), monounsaturated fatty acids (MUFA), and PUFA. Esterified PUFA is mainly affected by lipid peroxidation during ferroptosis. It is essential to consume sufficient amounts of unsaturated fatty acids to facilitate ferroptosis [
19]. How PUFAs are oxidized to produce peroxynitrite and trigger iron death should be further investigated. PUFA and MUFA have opposite roles in ferroptosis because of their differential susceptibility to oxidation [
18]. Ferroptosis is more susceptible to ferroptosis in the presence of increased PUFA synthesis, which is mainly controlled by ACSL4 and recombinant lysophosphatidylcholine acyltransferase 3. ACSL3 and stearyl coenzyme A dehydrogenase-1 (SCD-1) inhibit PUFA-dependent ferroptosis, respectively [
10,
20].
3.1. ACSL3 in Ferroptosis
ACSL3 may be involved in intracellular lipid metabolism by promoting LD formation and maturation. LDs are commonly found in the organelles of various cells, and they store neutral lipids, such as TAG and cholesteryl ester. ACSL3’s N-terminal domain controls the position of LD and FA absorption [
3]. ACSL3 grows LD; in the absence of LD, ACSL3 may be present in the ER and translocated to the LD surface during LD formation or after LD is reconnected to ER by membrane bridges [
21]. The transfer of ACSL3 from the ER to the LD occurs because of the strong affinity between ACSL3 and LD [
22]. Fujimoto et al. found that the addition of an oleic acid medium increased intracellular LD accumulation, which was accompanied by an increase in ACSL3 expression. Meanwhile, with the use of ACSL3 inhibitor triacsin C, the expression of the ACSL3 correspondingly decreased, accompanied by a significant decrease in intracellular LD content. This suggests that ACSL3 plays an important role in the process of ER budding LD growth and maturation after the cellular uptake of exogenous fatty acids [
23]. Further research has revealed that the mechanism of action may be that the LD-associated protein Rab18 (low molecular weight GTP-binding protein) interacts with perilipin 2 (PLIN2), a major LD protein, and forms a complex with PLIN2 and ACSL3 [
24].
Although the current study on ACSL3 is limited, some evidence shows that ACSL3 is highly relevant to ferroptosis and that ACSL3 expression is correlated with ferroptosis sensitivity [
5]. ACSL3 is essential for activating MUFA, which reduces the susceptibility of plasma membrane lipids to oxidation over a period of several hours. MUFA can modify cell membrane properties by replacing PUFAs. It inhibits iron-dependent oxidative cell death by preventing the accumulation of lipid ROS in the plasma membranes, and effectively inhibits the process of iron-dependent oxidative cell death. Based on the findings of Magtanong et al., treatment with exogenous MUFA reduces the sensitization of plasma membrane lipids to lethal oxidation within a few hours, and this process entails the activation of MUFA by ACSL3 [
12]. This study demonstrated that exogenous MUFAs and ACSL3 activities promote cellular resistance to ferroptosis. It has also been found that a combination of the anesthetic drug propofol or propofol injectable emulsion (PIE) and the chemotherapeutic drug paclitaxel inhibited cell viability and augmented the sensitivity of paclitaxel to cervical cancer cells. Mechanistically, ferroptosis-related pathways were influenced by drug treatments, including the SLC7A11/GPX4 ubiquinol/CoQ10/FSP1 and YAP/ACSL4/TFRC pathways [
25].
3.2. ACSL4 in Ferroptosis
As a specific biomarker and driver of ferroptosis, ACSL4 dictates ferroptosis sensitivity by altering the composition of cellular lipids [
13,
26]. Ferroptosis is induced by the iron-dependent peroxidation of lipids, mostly PLs containing PUFAs [
13]. ACSL4 is not only a sensitive monitor for ferroptosis but an essential regulator of lipid metabolism in ferroptosis. The ACSL4 gene-encoded enzyme has a strong preference for 20-carbon PUFA substrates, such as arachidonic acid (AA) and adrenaline (ADA), catalyzing their conversion to AA-CoA and ADA-CoA, producing LPO, and finally promoting ferroptosis [
27,
28]. Notably, oxidized AA and epinephrine phosphatidylethanolamines (Pes) have been proven to induce ferroptosis in cells [
29].
A mechanistic study showed that ACSL4 is a target gene of the oncoprotein YAP. Such an interaction between E-cadherin-mediated cell-cell contacts affecting the cell density on ferroptosis in epithelial cells could activate intracellular Hippo signaling via the NF2/merlin tumor suppressor, and thus inhibit the transcription of various target genes of YAP, including
ACSL4,
TfR1, and others [
30]. This discovery indicates that additional potential signaling pathways regulating ferroptosis by influencing ACSL4 levels are worthy of investigation and targeting these pathways may be useful in regulating cell death. An analysis revealed that ACSL4 and GPX4 are novel predictive and prognostic biomarkers for patients receiving neoadjuvant chemotherapy, which provided promising evidence for the regulation of ferroptosis in BC treatment [
31]. Currently, ACSL4 is studied in cervical cancer mostly in relation to drug therapy. Oleanolic acid (OA) is an anti-cancer compound found naturally in the leaves, fruits, and rhizomes of plants. Research has shown that OA activates ferroptosis in cervical cancer cells by enhancing ACSL4 expression and then a significant decrease in the viability and proliferative capacity of cancer cells was observed after exposure to OA [
32]. Curcumin, a yellow polyphenol compound derived from turmeric plants, has also been shown to induce ferroptosis with increasing levels of ACSL4, which presents a theoretical basis for the application of ferroptotic inducers in the treatment of lung cancer [
33]. Clinically, radiotherapy upregulates the expression of ACSL4, thereby increasing lipid synthesis and subsequent oxidative damage, thus inducing ferroptosis [
34]. In addition to its involvement in tumor progression, ACSL4 serves as a biomarker for sorafenib in the treatment of hepatocellular carcinoma (HCC) [
35]. The results of this study demonstrate that the ETS1/miR-23a-3p/ACSL4 axis contributes to sorafenib resistance in HCC by regulating ferroptosis. ACSL4 is upregulated in ovarian cancer (OC) and is a direct target of miR-424-5p, which inhibits ferroptosis in OC cells [
36]. The results of these studies have identified a potential therapeutic strategy for treating OC. These studies clearly indicate that there is a relationship between ACSL4 and ferroptosis in various cancers; however, more mechanisms among them remain to be further investigated and may contribute to more effective clinical treatments.