Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Endocrinology & Metabolism
Cardiac hypertrophy is a well-known condition that indicates the possibility of hypercortisolemia-inducing ACS. Because of its relationship with induction of heart failure and cardiac death, timely detection of such an alteration can lead to early diagnosis and contribute to reducing mortality.
- adrenal tumor
- autonomous cortisol secretion
- cardiac functional disorder
1. Glucocorticoid Receptor and Regulating Enzymes
The effects of bioactive cortisol, which is synthesized in the adrenal cortex, are exerted following connection to a glucocorticoid (GC) receptor (GR) or mineralocorticoid receptor (MR). GR belongs to a ligand-dependent transcription factor nuclear receptor superfamily that is comprised of three functional domains: an N-terminal transactivation domain (NTD), central DNA-binding domain (DBD), and C-terminal ligand-binding domain (LBD) [24]. Nearly all human tissues and organs express GR, which forms a multiprotein complex in cytoplasm [25]. Cortisol binding induces GR nuclear translocation with dissociation of the complex, including heat shock protein 90 [26], after which GC signaling enhances genomic and rapid non-genomic effects. MR is also a member of the ligand-dependent transcription factor nuclear receptor superfamily, showing a structural homology with GR, and induces genomic and rapid non-genomic effects in the same way as GR, though the outcome differs largely from that of GR activation and limited localization has been shown, such as cortical collecting ducts, epithelium of the large intestine and cerebrum, endothelium of blood vessels, and cardiac muscle tissue. Although both cortisol and aldosterone can function as a ligand to MR, it should be noted that cortisol has 100- to 1000-fold higher concentration in blood, and a 10- to 30-fold higher affinity to MR as compared with aldosterone. Furthermore, cortisol potentially shows both agonist and antagonist characteristics for MR [27]. However, MR transcription is seldom activated without stress or tissue injury [28,29,30], and GC was shown to function as an MR agonist in a rat heart failure model [31].
To produce an appropriate effect of cortisol and aldosterone, 11β-hydroxysteroid dehydrogenase type 1 and 2 (11β-HSD1, 2) play essential roles [32,33]. 11β-HSD1 converts inert 11-keto forms (cortisone, 11-dehydrocorticosterone) into cortisol by using the co-substrate NADPH provided by hexose-6-phophate dehydrogenase (H6PDH) [34], which results in an increase in local active cortisol concentration [35,36,37]. On the other hand, 11β-HSD2 inactivates cortisol [38,39], and has a high affinity but low capacity for NAD-dependent dehydration [40,41]. Therefore, the degree of the effect of cortisol on individual target cells is dependent on the balance of an intracellular GC-activating enzyme (11β-HSD1) and -inactivating enzyme (11β-HSD2) [42], with 11β-HSD1 inferior to 11β-HSD2 in regard to the strength of cortisol binding. For example, a suppressed effect of GC would be expected in the kidneys, where 11β-HSD2 is significantly expressed, unlike in the heart [28].
2. Physiological Role of Glucocorticoid in Rodent Cardiomyocytes
Endogenous GC contributes to maintain heart performance for regulating the life cycle of cardiomyocytes involved in growth, differentiation, metabolism, and apoptosis [43]. GC action via GR matures fetal cardiomyocytes and myofibrillar, and boosts its mitochondrial activation, by which contractile force is reinforced [44]. Narayanan et al. assessed the potential therapeutic benefits of dexamethasone treatment on myocardial function in senescent rats and demonstrated that it can reverse contractile performance by approximately two-fold caused by increased uptake of ATP-energized Ca2+ in the sarcoplasmic reticulum [45]. In addition, GC potentially inhibits cardiomyocyte apoptosis [46,47] by activating serum and glucocorticoid-responsive kinase (SGK-1), B-cell lymphoma-extra large (Bcl-xL), and growth arrest specific 2 (Gas2) [48,49]. Additionally, GR potentially protects cardiomyocytes from DNA damage and drug-induced cell death by regulating the expression of Kruppel-like factor 13 (KLF13), a major mediator of GR [50].
H6PDH activity has been identified in cardiomyocytes and fibroblasts in rats [51,52], while HSD11B1 mRNA has been observed in human hearts [53], though a physiologically normal condition restricts GC regeneration [54,55,56]. Nevertheless, 11β-HSD1 is essential for maintaining contractile force generation [54] and heart growth [57]. Interestingly, White et al. reported that cardiomyocytes lacking 11β-HSD1 in perinatal mice matured with a shortened length, though a phenotypically normal heart was successfully developed in those animals [58]. In relation to these findings, Rahman et al. presented the possibility of a reduction in left ventricular mass caused by a single nucleotide polymorphism in the HSD11B1 gene [59].
3. Harmful Effects to Cardiomyocytes by Experimental Excessive Glucocorticoid Exposure
3.1. Excessive Glucocorticoid and Cardiomyocyte Hypertrophy
Regarding the effects of excessive GC in therapeutic and pathologic conditions, abundant knowledge showing its influence with exposure to the heart has been accumulated. For example, antenatal corticosteroid therapy, known to reduce neonatal death [60], has been found to have a limited contribution to preterm birth [61], whereas it is associated with high risks of cardiovascular disease, hypertension, and type 2 diabetes in adults [62,63,64,65,66,67]. Ren et al. explored the direct effects of excessive GC on cardiomyocytes by exposing a rat embryonic cardiomyocyte cell line (H9C2) and primary cardiomyocytes to dexamethasone [50]. The results showed a significant increase in cell size along with elevated levels of expression of cardiomyocyte hypertrophic markers, such as atrial natriuretic factor (ANF), β-myosin heavy chain (β-MHC), and skeletal muscle α-actin (α-SKA) (Figure 1). Moreover, Ingenuity Pathway Analysis revealed that 58 genes were associated with cardiomyocyte hypertrophy signaling at 48 h after dexamethasone treatment. Additionally, dexamethasone was found to exhibit an anti-apoptotic effect on cardiomyocytes with exposure to tumor necrosis factor (TNF)-α with serum deprivation, while its deprivation abolished the effect of dexamethasone to elevate the expression levels of those hypertrophic maker genes, except for β-MHC. These results were diminished by addition of a GR antagonist or knock-down of GR expression, while suppression of MR activity did not have such effects, indicating the essential relationship of GR activity. Lister et al. presented findings that verified altered corticosteroid signaling in cardiomyocyte hypertrophy induced by phenylephrine (α-adrenergic receptor agonist) and phorbol ester (protein kinase C (PKC) activator), and as such signal activation is known to be involved in hypertension and diabetes [68]. Consequently, a hypertrophic response was found to accompany a significant increase in atrial natriuretic peptide mRNA (8- to 12-fold increase in both) and rDNA transcription (2-fold increase), which exhibit corticosteroid effects, and also GR and MR expression (2-fold increase) with enhanced receptor signaling. When priming with phenylephrine was performed, corticosteroids potentiated a hypertrophic response via GR, while phorbol ester-induced hypertrophy exhibited increased 11β-HSD1 expression and its reductase activity. These results indicate cross talk between corticosteroid receptor-activated pathways, and both α-adrenergic and PKC signals (Figure 1).
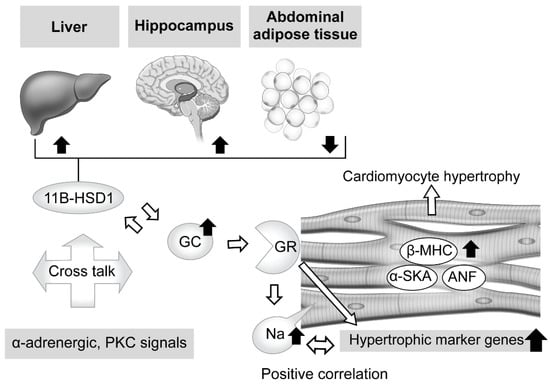
Figure 1. Effects of excessive glucocorticoid in cardiomyocytes and modulators of its signaling, including tissue-dependent regulation of 11β-hydroxysteroid dehydrogenase type 1. Excessive glucocorticoid (GC) enlarged the cell size of rat cardiomyocytes, with elevation of cardiomyocyte hypertrophic markers and related genes, such as atrial natriuretic factor (ANF), β-myosin heavy chain (β-MHC), and skeletal muscle α-actin (α-SKA). The glucocorticoid receptor (GR) antagonist and knockdown of GR expression attenuated that reaction, whereas suppression of mineralocorticoid receptor (MR) activity did not, indicating an essential role for GR in such a response. Phenylephrine (α-adrenergic receptor agonist) and phorbol ester (protein kinase C (PKC) activator), known to induce cardiomyocyte hypertrophy and related to hypertension and diabetes, potentiate the effects of corticosteroid and increase activated GR and MR expression. In addition, phorbol ester-induced hypertrophy exhibited increased 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) expression and reductase activity. These results indicate possible cross talk between corticosteroid receptor-activated pathways, and both α-adrenergic and PKC signals. Corticosteroid also induces an increase in intracellular sodium concentration, which is related to ischemia, heart failure, and hypertrophy. That increase was revealed to have a positive correlation with hypertrophic gene expression. Furthermore, excessive corticosterone increased the expression level of 11β-HSD1 mRNA in abdominal adipose tissue, while that was decreased in the hippocampus and liver, suggesting regulation in a tissue-dependent manner.
On the other hand, intracellular sodium kinetics in ischemia, heart failure, and hypertrophy has been considered [69,70,71,72,73]. According to a report by Katoh et al., cultured neonatal rat ventricular myocytes were treated for 24 h with corticosterone, aldosterone, and dexamethasone, and the results showed a nearly 1.5-fold increase in intracellular sodium concentration, though that occurred in a dose-dependent manner [74]. In addition, a GR antagonist reduced intracellular sodium concentration, and positive correlations between hypertrophic gene expressions and concentration were observed, findings not obtained with an MR antagonist. Additional experiments then revealed that dexamethasone upregulated the mRNA of sodium-calcium exchanger (NCX)1 and its protein, which is related to intracellular sodium homeostasis and influences calcium efflux in cardiomyocytes [72]. These findings suggest that exposure to excessive GC increases the concentration of intracellular sodium via GR, while dexamethasone treatment explains, at least in part, the direct effect of that increase, leading to cardiomyocyte hypertrophy (Figure 1).
3.2. Tissue-Specific Role of 11β-Hydroxysteroid Dehydrogenase Type 1 in Glucocorticoid Excess
When considering local regulation of GC, its association with 11β-HSD1 is another related issue. Nishiyama et al. recently confirmed the influence of 11β-HSD1 in mouse tissues showing persistent GC excess [75]. Two weeks of administration of excessive corticosterone to male mice decreased expression levels of 11β-HSD1 mRNA in the hippocampus and liver, while those were increased in abdominal adipose tissue. Furthermore, similar results were obtained with male corticotropin releasing hormone (CRH)-overexpressing transgenic mice, an animal model of CD [76,77], while performance of an adrenalectomy reversed those changes, suggesting a tissue-dependent action (Figure 1). Huang et al. also noted the role of 11β-HSD1 in cardiomyocytes [78]. Briefly, treatment of primary neonatal rat ventricular cardiomyocytes (NRCMs), which show upregulated 11β-HSD1 expression, with palmitic acid induced a significant enlargement of cell size and increased the mRNA levels of cardiomyocyte hypertrophy-specific genes, including ANF, SKA, and β-MHC, whereas either a selective inhibitor of 11β-HSD1-treated or 11β-HSD1-deficient NRCMs caused a decrease in cell size. Moreover, they also confirmed marked attenuation of 11β-HSD1-induced hypertrophy of cardiomyocytes in not only the presence of a GR antagonist (RU486), but also with the MR antagonist spironolactone, which inhibits nuclear MR translocation [79].
This entry is adapted from the peer-reviewed paper 10.3390/jcm11237035
This entry is offline, you can click here to edit this entry!