Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is an autoimmune liver disease that mostly affects women. A progressive disorder in the processes of bile secretion and enterohepatic bile salts circulation in patients with PBC already in its early stages, leading to an insufficient release of bile acids into the bowel and their entry into the systemic circulation. Insufficient bile acids released into the duodenum contributes to the development of malabsorption, energy malnutrition, and slowly progressive weight loss.
- primary biliary cholangitis (PBC)
- dietary lipid metabolism disorders in PBC
1. Introduction
Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is a chronic progressive cholestatic granulomatous and a destructive inflammatory lesion of small intralobular and septal bile ducts, which is likely to be caused by an autoimmune mechanism in the presence of serum antimitochondrial antibodies and a potential tendency to progress to cirrhosis [1,2,3]. Autoimmune tolerance defects are critical factors in disease initiation and the gradual development of intrahepatic cholestasis [3,4]. The latter in PBC is associated with damage to subcellular structures in the intrahepatic bile duct epithelial cells and with a change in the metabolism of bile acids due to the disrupted processes of bile secretion and their enterohepatic circulation. Progressive intrahepatic cholestasis results in an insufficient release of bile acids into the duodenum and, on the other hand, their increased accumulation in the hepatocytes and plasma. It is precisely these bile production and excretion changes that should be considered as an underlying cause of lipid metabolism disorders in PBC. In this case, lipid metabolism and transport disorders occur with dietary lipids as well as lipids and their transporting systems synthesized in the body.
2. The Mechanism of Dietary Lipid Metabolism Disorders in PBC
Insufficient release of bile acids into the duodenum in patients with PBC leads to a decrease in the rate of hydrolysis of fats and a reduction in the absorption of fats and fat-soluble vitamins. This contributes to the development of steatorrhea, protein–energy, and vitamin–mineral malnutrition. [5,6,7,8]. The seriousness of steatorrhea correlates with the decreased excretion and concentration of bile acids in the intestinal lumen (r = 0.82; p < 0.0001), with the level of a serum bilirubin increase (r = 0.88; p < 0.001), and with the late histological stages of PBC (p < 0.005) [9]. Insufficient emulsification of fats underlies the mechanism of the development of steatorrhea [6]. At the same time, the exocrine function of the pancreas is not disturbed. The lipases synthesized by the pancreas are involved in the hydrolysis of insufficiently emulsified fats, which slows down the rate of their splitting. The results obtained by Ross et al. indicate that pancreatic synthesis of lipases and their entry into the duodenum in PBC is not impaired and is not the cause of steatorrhea development [10]. The activity of pancreatic amylase in patients with PBC is within the normal range [10,11]. The severity of steatorrhea also does not correlate with the increased activity of alkaline phosphatase in these patients [10,11].
In the intestine, bile acids are taken part in the absorption of fat-soluble vitamins and fat hydrolysis products. Monoglycerides and fatty acids formed as a result of the hydrolysis of triglycerides and phospholipids, as well as fat-soluble vitamins together with bile acids, form lipoid-bile complexes that are absorbed by enterocytes (Figure 1). The latter are formed due to the detergent properties of bile acids, which are able to solubilize fatty acids and monoglycerides with the formation of micellar or lamellar structures [5,7,8]. Inside the enterocytes, the complexes break down. Fatty acids are used by the enterocytes as a building and/or energy material, or together with tri-, di-, and mono-glycerides and phospholipids form chylomicrons for further transport through the body. At the same time, bile acids again can be released into the intestinal lumen and take part in the processes of emulsification and absorption of fats and fat-soluble vitamins. This process can be repeated 4–6 times during the passage of bile acids through the intestine [12].
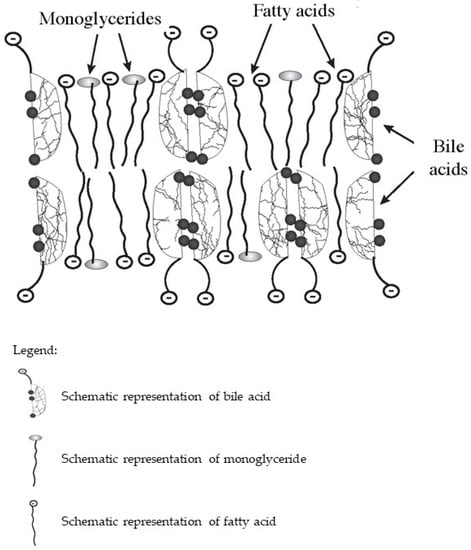
Figure 1. Schematic diagram of the composition of lipoid-bile complexes formed in the small bowel.
The insufficient release of primary bile acids in the intestine in PBС leads to a change in the composition of the microbiome, causing gut dysbiosis [10,13,14,15]. According to DiBaise et al., dysbiosis in patients with PBC also determines the development of steatorrhea [16]. Therefore, these patients should be necessarily evaluated for bacterial overgrowth [16].
Since insufficient bile acid release into the bowel is one of the first signs, impurities of partially digested fecal fats can be detected in patients with PBC already at an early stage. Steatorrhea in PBC is manifested with diarrhea varying in severity. At the same time, some patients with PBC may experience constipation. The development of the latter may be associated with a deficiency of bile acids that stimulate intestinal motility and gut dysbiosis [13,17,18].
Gradually and imperceptibly progressing steatorrhea leads to malnutrition and slowly progressive weight loss in patients with PBC [8], which is manifested only by general weakness and/or lower performance for quite a long time [19,20,21]. Even a slight nutrient deficiency is accompanied by gradually progressing glycogenolysis and a reduction in glycogenogenesis, which results in the activation of compensatory mechanisms. The latter is aimed at protecting vital organs that need higher energy consumption [22]. As a result, reserves of adipose tissue are used as an energy material. The use of fatty acids as an energy material and activation of β-oxidation of fatty acids is accompanied by the development of slowly progressive weight loss and malnutrition in patients with PBC [8,23].
3. Features of Lipid Metabolism Disorders in PBC
Due to the developing and increasing cholestasis, bile acids is accumulated in hepatocytes and enter them into the blood plasma. The appearance of bile acids in the general bloodstream leads to the development of dyslipidemia. The plasma levels of palmitic and oleic acids, as well as PhLs and cholesterol, increase in patients with PBC already in its early stages [5,66]. Some authors have primarily attempted to consider the detectable increase in TC in PBC patients in terms of its effect on the development of atherosclerosis and pathological cardiovascular changes in these patients [32,43,67]. However, the elevated levels of cholesterol in patients with PBC, as well as those of phospholipids, are aimed at neutralizing the detergent effect of bile acids that have entered the systemic circulation as cholestasis progresses. In this regard, dyslipidemia in PBC is “anomalous” and is the body’s compensatory response to the appearance of bile acids in systemic circulation. Therefore, despite the higher plasma TC levels in PBC patients, the latter is found in elevated HDL concentrations, poses a low risk for atherosclerosis and cardiovascular events, has a low-grade development of hepatic steatosis, and has the appearance of abnormal plasma Lp-X [25,31,67,68]. In this regard, PBC can serve as a model system for developing a new area in the search and design of drugs to treat dyslipidemia.
The studies conducted by Y. Zhang et al. show that patients with PBC have the lowest degree of hepatic steatosis not only among those with chronic liver diseases, but also have a lower degree than in healthy people [25]. The mechanism of low-grade concomitant hepatic steatosis is multifactorial. Insufficient intestinal bile acids release leading to fat malabsorption, steatorrhea, and gut microbial dysbiosis in PBC patients can cause a decrease in liver fat deposition [5,8]. In addition, the higher expression of FGF 19 induces a reduction in mitochondrial acetyl-coenzyme A-carboxylase 2, promoting free fatty acid oxidation and simultaneously inhibiting fatty acid synthesis, which lowers liver fat accumulation and plasma triglyceride levels [70].
The presence of Lp-X in cholestatic liver diseases is of great clinical importance, since its detection is considered to be the most sensitive and specific biochemical marker of cholestasis [45]. A positive Lp-X test shows more than 95% agreement with histological methods used to confirm cholestatic syndrome [71]. However, due to the complexity of determining Lp-X and the high information value of biochemical markers, the determination of alkaline phosphatase, and γ-glutamyl transferase, a test for Lp-X is rarely used to diagnose the cholestatic state.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10123046
This entry is offline, you can click here to edit this entry!