Alzheimer’s disease (AD) is a hereditary and sporadic neurodegenerative illness defined by the gradual and cumulative loss of neurons in specific brain areas. The processes that cause AD are still under investigation and there are no available therapies to halt it. Progress puts at the forefront the “calcium (Ca2+) hypothesis” as a key AD pathogenic pathway, impacting neuronal, astrocyte and microglial function. An increasing body of evidence points out the early and crucial role of cellular Ca2+ handling dysregulation in AD pathogenesis. Interestingly, Ca2+ is a key regulator of several mitochondrial functions, such as ATP production, and brain cells rely mostly on OXPHOS to match their energy demands.
- calcium
- Alzheimer’s disease
- mitochondria
- bioenergetics
- neuron
- microglia
- astrocyte
- AD mouse model
- iPSCs
1. Introduction
2. The Physiology of Brain Mitochondria: Ca2+ and Bioenergetics
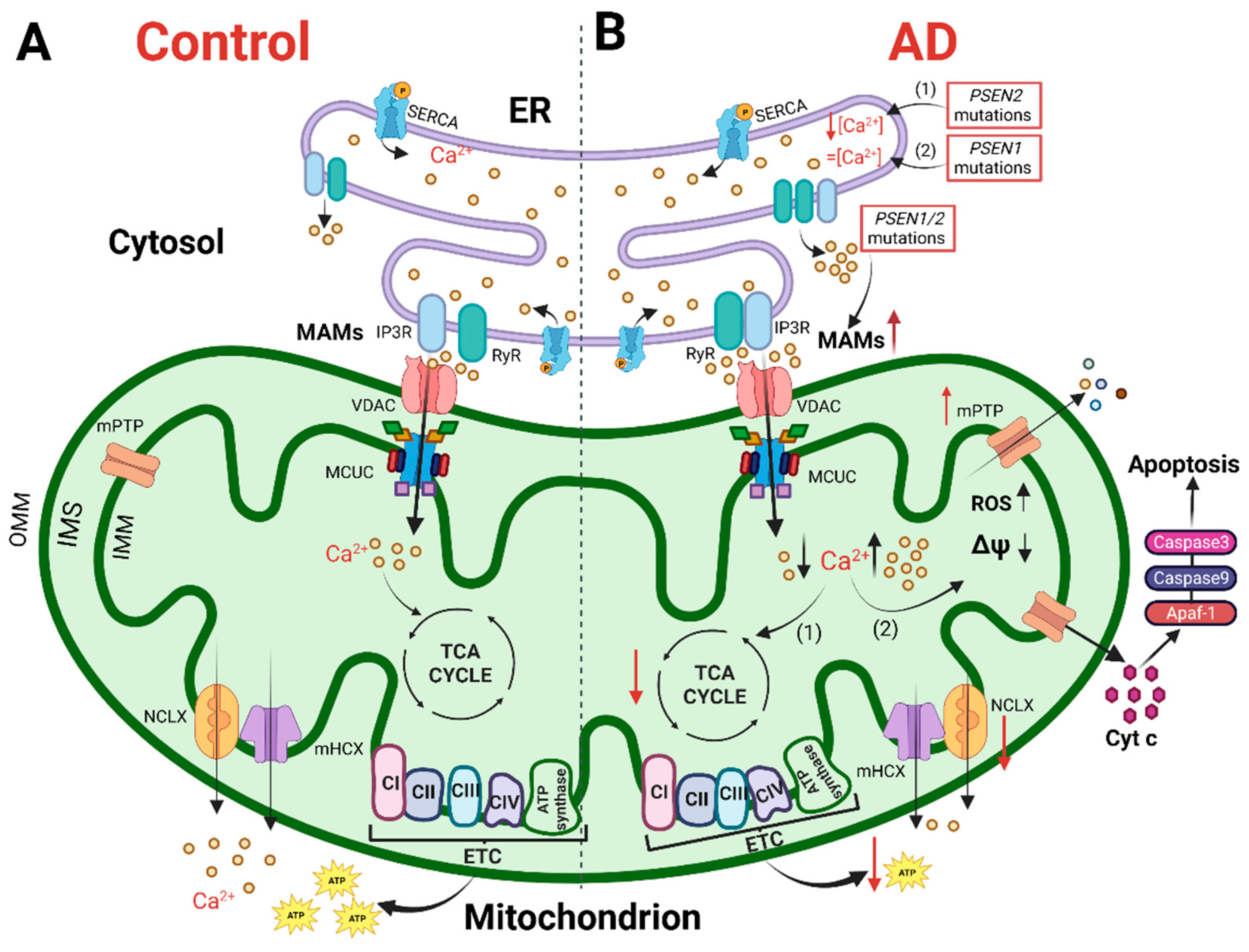
3. Mitochondria in Alzheimer’s Disease: Ca2+ Signaling and Bioenergetics
4. Mitochondria Ca2+ Signaling and Bioenergetics as Therapeutic Targets
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10123025
References
- Sokoloff, L. Energetics of functional activation in neural tissues. Neurochem. Res. 1999, 24, 321–329.
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019.
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51.
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460.
- Niu, H.; Álvarez-Álvarez, I.; Guillén-Grima, F.; Aguinaga-Ontoso, I. Prevalence and incidence of Alzheimer’s disease in Europe: A meta-analysis. Neurologia 2017, 32, 523–532.
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868.
- Altomari, N.; Bruno, F.; Laganà, V.; Smirne, N.; Colao, R.; Curcio, S.; Di Lorenzo, R.; Frangipane, F.; Maletta, R.; Puccio, G.; et al. A Comparison of Behavioral and Psychological Symptoms of Dementia (BPSD) and BPSD Sub-Syndromes in Early-Onset and Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 691–699.
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517.
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185.
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048.
- Khan, A.; Corbett, A.; Ballard, C. Emerging treatments for Alzheimer’s disease for non-amyloid and non-tau targets. Expert Rev. Neurother. 2017, 17, 683–695.
- Poudel, P.; Park, S. Recent Advances in the Treatment of Alzheimer’s Disease Using Nanoparticle-Based Drug Delivery Systems. Pharmaceutics 2022, 14, 835.
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2.
- Bruno, F.; Malvaso, A.; Canterini, S.; Bruni, A.C. Antimicrobial Peptides (AMPs) in the Pathogenesis of Alzheimer’s Disease: Implications for Diagnosis and Treatment. Antibiotics 2022, 11, 726.
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63.
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321.
- Callens, M.; Loncke, J.; Bultynck, G. Dysregulated Ca2+ Homeostasis as a Central Theme in Neurodegeneration: Lessons from Alzheimer’s Disease and Wolfram Syndrome. Cells 2022, 11, 1963.
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166.
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215.
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61, 24–36.
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430.
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174.
- Zhang, Q.; Sidorenko, J.; Couvy-Duchesne, B.; Marioni, R.E.; Wright, M.J.; Goate, A.M.; Marcora, E.; Huang, K.L.; Porter, T.; Laws, S.M.; et al. Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat. Commun. 2020, 11, 4799.
- Wightman, D.P.; Jansen, I.E.; Savage, J.E.; Shadrin, A.A.; Bahrami, S.; Holland, D.; Rongve, A.; Børte, S.; Winsvold, B.S.; Drange, O.K.; et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 2021, 53, 1276–1282.
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30.
- Cavalcante, G.C.; Brito, L.M.; Schaan, A.P.; Ribeiro-Dos-santos, Â.; de Araújo, G.S. Mitochondrial Genetics Reinforces Multiple Layers of Interaction in Alzheimer’s Disease. Biomedicines 2022, 10, 880.
- Flaquer, A.; Baumbach, C.; Kriebel, J.; Meitinger, T.; Peters, A.; Waldenberger, M.; Grallert, H.; Strauch, K. Mitochondrial genetic variants identified to be associated with BMI in adults. PLoS ONE 2014, 9, e105116.
- Rumshisky, A.; Ghassemi, M.; Naumann, T.; Szolovits, P.; Castro, V.M.; McCoy, T.H.; Perlis, R.H. Predicting early psychiatric readmission with natural language processing of narrative discharge summaries. Transl. Psychiatry 2016, 6, e921.
- Schröder, H.; Moser, N.; Huggenberger, S. Neuroanatomy of the Mouse; Springer International Publishing: Cham, Switzerland, 2020.
- de Rus Jacquet, A.; Denis, H.L.; Cicchetti, F.; Alpaugh, M. Current and future applications of induced pluripotent stem cell-based models to study pathological proteins in neurodegenerative disorders. Mol. Psychiatry 2021, 26, 2685–2706.
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117.
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476.
- Perrone, M.; Caroccia, N.; Genovese, I.; Missiroli, S.; Modesti, L.; Pedriali, G.; Vezzani, B.; Vitto, V.A.M.; Antenori, M.; Lebiedzinska-Arciszewska, M.; et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. In International Review of Cell and Molecular Biology; Elsevier Publighing: Amsterdam, The Netherlands, 2020; ISBN 9780128197448.
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 2017, 358, 623–630.
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610.
- Ooi, K.; Hu, L.; Feng, Y.; Han, C.; Ren, X.; Qian, X.; Huang, H.; Chen, S.; Shi, Q.; Lin, H.; et al. Sigma-1 Receptor Activation Suppresses Microglia M1 Polarization via Regulating Endoplasmic Reticulum–Mitochondria Contact and Mitochondrial Functions in Stress-Induced Hypertension Rats. Mol. Neurobiol. 2021, 58, 6625–6646.
- Göbel, J.; Engelhardt, E.; Pelzer, P.; Sakthivelu, V.; Jahn, H.M.; Jevtic, M.; Folz-Donahue, K.; Kukat, C.; Schauss, A.; Frese, C.K.; et al. Mitochondria-Endoplasmic Reticulum Contacts in Reactive Astrocytes Promote Vascular Remodeling. Cell Metab. 2020, 31, 791–808.e8.
- Khachaturian, Z.S. Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol. Aging 1987, 8, 345–346.
- Khachaturian, Z.S. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann. N. Y. Acad. Sci. 1989, 568, 1–4.
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872.
- Landfield, P.W.; Pitler, T.A. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science 1984, 226, 1089–1092.
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20.
- Filadi, R.; Greotti, E. The yin and yang of mitochondrial Ca2+ signaling in cell physiology and pathology. Cell Calcium 2021, 93, 102321.
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166.
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Mol. Cell 2017, 67, 711–723 e7.
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045.
- Di Marco, G.; Vallese, F.; Jourde, B.; Bergsdorf, C.; Sturlese, M.; De Mario, A.; Techer-Etienne, V.; Haasen, D.; Oberhauser, B.; Schleeger, S.; et al. A High-Throughput Screening Identifies MICU1 Targeting Compounds. Cell Rep. 2020, 30, 2321–2331.e6.
- De Mario, A.; Tosatto, A.; Hill, J.M.; Kriston-Vizi, J.; Ketteler, R.; Vecellio Reane, D.; Cortopassi, G.; Szabadkai, G.; Rizzuto, R.; Mammucari, C. Identification and functional validation of FDA-approved positive and negative modulators of the mitochondrial calcium uniporter. Cell Rep. 2021, 35, 109275.
- Montero, M.; Lobaton, C.D.; Hernandez-Sanmiguel, E.; Santodomingo, J.; Vay, L.; Moreno, A.; Alvarez, J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem. J. 2004, 384, 19–24.
- Bermont, F.; Hermant, A.; Benninga, R.; Chabert, C.; Jacot, G.; Santo-Domingo, J.; Kraus, M.R.C.; Feige, J.N.; De Marchi, U. Targeting Mitochondrial Calcium Uptake with the Natural Flavonol Kaempferol, to Promote Metabolism/Secretion Coupling in Pancreatic β-cells. Nutrients 2020, 12, 538.
- Rodríguez, L.R.; Lapeña-Luzón, T.; Benetó, N.; Beltran-Beltran, V.; Pallardó, F.V.; Gonzalez-Cabo, P.; Navarro, J.A. Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases? Antioxidants 2022, 11, 165.
- Neumann, J.T.; Diaz-Sylvester, P.L.; Fleischer, S.; Copello, J.A. CGP-37157 inhibits the sarcoplasmic reticulum Ca2+ ATPase and activates ryanodine receptor channels in striated muscle. Mol. Pharmacol. 2011, 79, 141–147.
- Onyango, I.G. Modulation of mitochondrial bioenergetics as a therapeutic strategy in Alzheimer’s disease. Neural Regen. Res. 2018, 13, 19–25.
- De La Rossa, A.; Laporte, M.H.; Astori, S.; Marissal, T.; Montessuit, S.; Sheshadri, P.; Ramos-Fernández, E.; Mendez, P.; Khani, A.; Quairiaux, C.; et al. Paradoxical neuronal hyperexcitability in a mouse model of mitochondrial pyruvate import deficiency. Elife 2022, 11, e72595.