Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Aspirin (ASA, acetylsalicylic acid ATC code: N02BA01, DrugBank ID: DB00945, brand names: Arthritis Pain, Aspi-Cor, Aspirin 81, Aspirin-Low, Bayer Plus, Bufferin, Ecortin, Eciprin, Miniprin, Vazalore), is one of the first drugs to be obtained by synthesis. It is regarded as, being the most used drug with the longest lasting commercial success.
- aspirin (ASA)
- ASA formulations
- ASA derivatives
1. Introduction
Aspirin (ASA, acetylsalicylic acid ATC code: N02BA01 [1], DrugBank ID: DB00945 [2], brand names: Arthritis Pain, Aspi-Cor, Aspirin 81, Aspirin-Low, Bayer Plus, Bufferin, Ecortin, Eciprin, Miniprin, Vazalore [3]), is one of the first drugs to be obtained by synthesis. It is regarded as, being the most used drug with the longest lasting commercial success [4]. ASA was originally used predominantly as an anti-inflammatory medication [5]. Nowadays, ASA is still a favorite of patients with a consumption of 44,000 tons of ASA each year, which is equivalent to approximately 120 billion aspirin tablets [6].
So far, there have been a significant number of publications written about ASA—researchers have focused on the uniqueness of the structure—and want to bring it closer to the reader. The use of aspirin-like drugs in modern medicine is very broad and includes the treatment of inflammation, pain and various cardiovascular diseases [7]. Many studies show that the benefits of using ASA far outweigh the potential risk of side effects. Like other medicines, ASA is toxic in high doses (over 150 milligrams per kilogram of body weight) [8].
2. History of ASA
The first use of willow bark containing salicin as analgesic was described in the Ebers Papyrus in 1534 BC [9]. Hippocrates also used an infusion of willow bark to treat pain. The key precursor for the synthesis of ASA was salicylic acid (SA). SA was chemically described and synthesized in 1859 by Hermann Kolbe [10][11]. However, the pharmacological use of SA was significantly limited due to its side effects such as nausea, gastric irritation and tinnitus. An important step in history of ASA was first synthesis of pure and stable form achieved by Felix Hoffmann [12]. The date of this synthesis (10 August 1897) is considered as the birthday of ASA [13]. Sales of ASA in tablet form began in 1904 and contributed to immediate commercial success of these drugs. It is worth emphasizing that ASA is one of the first industrial drugs available in the form of tablets in the world. On 1 February 1899, Bayer registered the trademark name in Berlin. ASA was then patented (patent no. 644077) in the United States in 1900. After this, ASA started its great triumph and became the most popular painkiller worldwide [14]. Already in 1904, the annual production of ASA was 25,823 kg. The outbreak of World War I in 1914 interrupted the international trade in pharmaceuticals and caused the ASA to become available without prescription [15]. From this time, the history of ASA has been very rich. It seems that the breakthrough for this drug was the discovery of its anticoagulant activity. Lawrence Craven used ASA in primary cardiovascular prevention in 1953. ASA’s mechanism of action was discovered in 1971 by Vane, Samuelsson and Bergström who then received the Nobel Prize for the work [14][16].
A cursory search of the PubMed database reveals that 70,694 articles about ASA have been published, while Google Scholar indicates 1,360,000 scientific and medical papers on ASA. Nonetheless, ASA is still a very attractive drug for scientists who are looking for an antidote to new threats [17]. Even today, it is one of the most studied drugs in the world [18]. The U.S. Food and Drug Administration (FDA) Clinical Trials Register (CTR) databased found 2287 studies [19], while the EU CTR currently contains 431 clinical trials for ASA with the EudraCT protocol, of which 69 are clinical trials conducted in people under 18 years of age [20], and the Australian and New Zealand CTR databases found only 87 studies [21]. As mentioned above, another important factor is that this substance is produced in great amount, i.e., in 2020 year it synthesized approx. 44,000 tons of ASA [6]. In the United States alone, more than 50 million people regularly take 10 to 20 billion ASA tablets to help prevent cardiovascular disease (CAVDs) and celebrovascular disease (CEVDs) [6][22].
3. ASA Structure
A total of 30 crystal structures of ASA are deposited in the Cambridge Structural Database (CSD) [23]. They have been assigned as ref codes from ACSALA to ACSALA29. The first crystal structure was obtained in 1964 [24] and was subsequently confirmed with greater accuracy about 20 years later [25] (Figure 1). ASA is O-acetyl (Ac) derivative of SA [26]. ASA has three bonds about which rotation is possible [27]. It was proven in computational study that form I has the lowest energetic minima [26]; due to this fact, it is the most stable ASA structure [28].
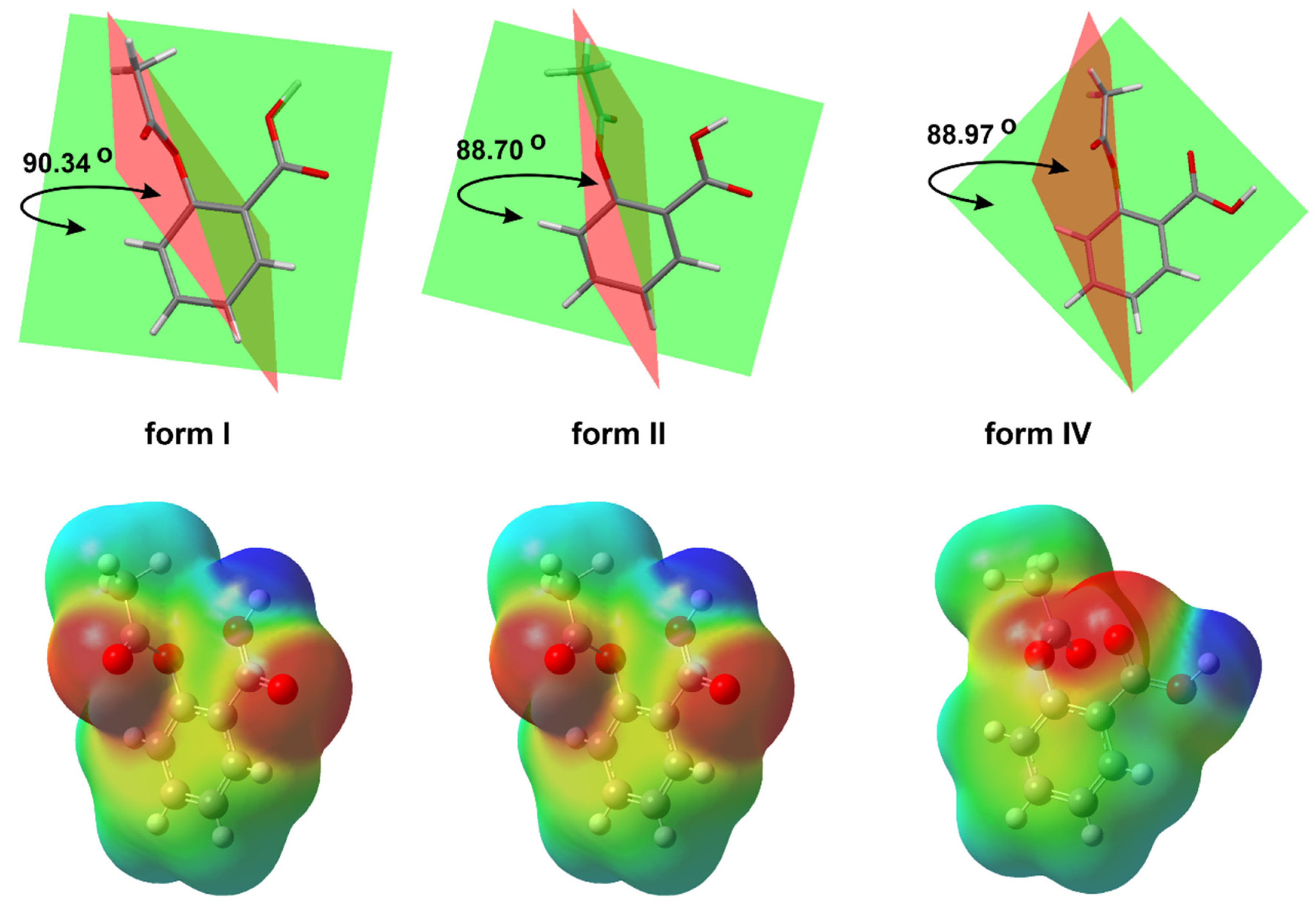
Figure 1. The basic crystal structure of ASA (CSD refcode ACSALA); compared 3D molecular view of the forms, I (CSD refcode ACSALA), II (CSD refcode ACSALA13) and IV (CSD refcode ACSALA24); the orientation of OH moiety with respect to the Ac group is on the near side in form I and II, but in form IV, the inverse arrangement of mentioned moieties can be observed; calculated molecular electrostatic potential map of ASA forms.
The phenomenon referred to as polymorphism relates to an organic molecule that can crystallize in more than one way or form [27][28][29][30]. Nowadays, it is assumed that ASA has four polymorphs [31]. From the pharmaceutical industry perspective, polymorph control is a very important issue due to the fact that each crystal form has different physical and chemical features, such as thermodynamic, kinetic, packing, surface, mechanical and spectroscopic properties. It determines the solubility, stability and bioavailability of the selected chemical as a drug [32]. In this way, the polymorphism directly affects the performance and functionality of the active ingredient in the drug product. The first crystal structure of form II was obtained in 2005 [33] (Figure 2).
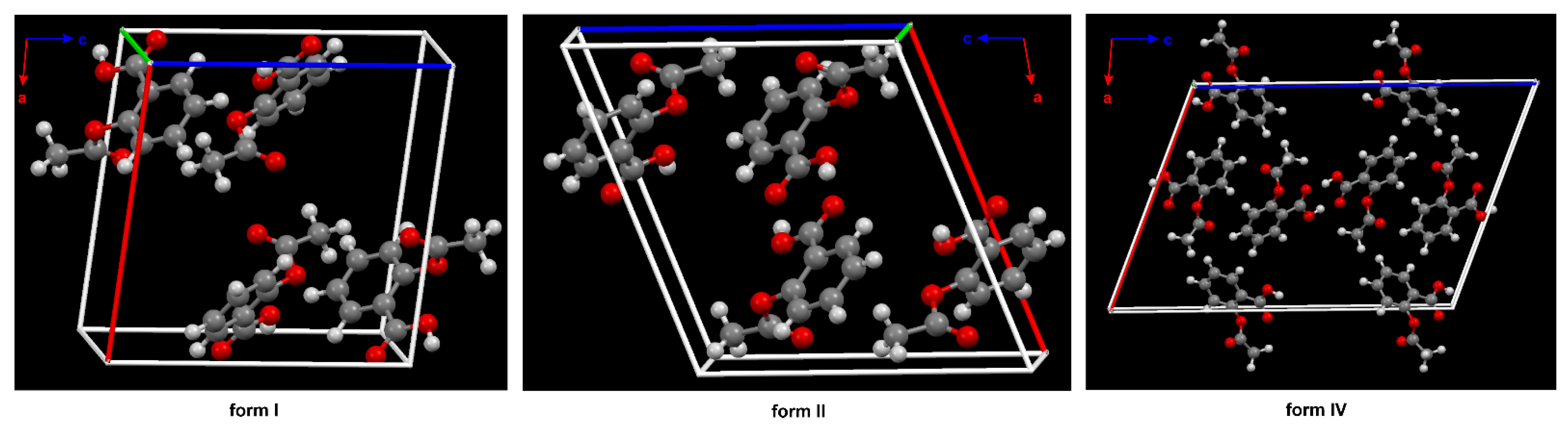
Figure 2. The crystal packing of ASA, form I (CSD refcode: ACSALA), form II (CSD refcode: ACSALA13) and form IV (CSD refcode: ACSALA24) along the b axis; formed through O–H ···O hydrogen bonds. The key difference between the structures I and II lies in the way the layers are arranged and bonded. Form I shows molecules in direct contact across the layer boundary forming the hydrogen bonded centrosymmetric acetyl groups (Ac) dimer. Form II shows the contraccatemericCO2H dimer and interlayer acid dimers are connected via catemeric methyl C–H ···O and phenyl C–H ···O hydrogen bonds interactions. The two arrangements are related to each other by a relative shift of adjacent layers along the crystallographic c axis in space group P21/c. The known crystal structure of ASA. In the structure of form IV, the plane of the Ac group is nearly perpendicular to the aryl ring plane, as in form I and II (see Figure 1). Color code: H = white, C = grey, O = red.
The form III was crystallized from form I through the transformation at high pressures (2 GPa) and was identified by means of Raman spectroscopy in 2015 [34]. Nevertheless, ASA crystal form III has not been obtained so far. Additionally, upon release of the pressure, form III transforms back to form I, indicating that the high-pressure phase is not stable at ambient conditions. Form IV was crystallized from the melt, and its structure was determined using a combination of X-ray powder diffraction analysis and crystal structure prediction algorithms in 2017 [35]. Optimized structure of ASA form IV (CSD refcode: ACSALA24) indicates that the orientation of the OH group of the CO2H moiety is on the far side with respect to the Ac group (Figure 1) [35]. The computational studies have suggested n → π* interaction between the aromatic COOH and the -C=O carbon of the Ac group [36]. Even after the discovery of the crystalline structure of ASA form II, the existence of the polymorph is still controversial due to the unique mutual growth phenomenon observed between the two polymorphic domains. However, it is clear that the two polymorphs exhibit significantly different solid state properties such as dissolution rate, mechanical properties, crystal habit, melting point and pKa [37].
3.1. The Basic Physical, Chemical and Biological Properties of ASA
Transport of pharmaceuticals in biologic tissues includes solvation in and distribution between environments of different properties in terms of lipophilicity, basicity, etc. [38][39][40]. The drug solubility testing in solvents present in the body is necessary to determine bioavailability, and the dose of the drug that should be administered to produce a specific therapeutic effect. The solubility of drugs depends on the pH of the environment in which the drug is absorbed and on the dissociation coefficient-pKa [41]. The solubility of a biologically active compound in water, i.e., in the main component of every organism, is checked for almost every compound. Other important solvents are ethanol (a model responsible for the transport of the drug in the body) and octanol (a model lipid, a component of biological cell membranes). SA is among the drugs that have been best studied in terms of their solubility in both organic and inorganic solvents [42]. The solubility of this drug was tested in such solvents as water, methanol or acetic acid. SA is poorly soluble in water. However, its sodium salt dissolves very well. ASA is more soluble in ethanol, ethyl ether, chloroform, sodium hydroxide solution and sodium carbonate solution than in water. However, with the access of moisture, it undergoes hydrolysis to SA and AcOH. It easily decomposes in aqueous solutions, and an increase in pH significantly accelerates both dissolution of the compound in water and its decomposition [39]. Table 1 presents the basic chemical, physical and biological properties of ASA compared to selected non-steroidal anti-inflammatory drugs (NSAIDs).
Table 1. The basic physical, chemical and biological properties of ASA compared to selected NSAIDs.
| Solvent | Solubility of ASA at 298 K, g/100 g [a] | Solubility of SA at 298 K, g/100 g | pKa | IC50 * | ||
|---|---|---|---|---|---|---|
| COX-1 | COX-2 | |||||
| H2O | 0.46 [d,e] | 0.22 [f] | ASA | 3.5 | 1.67 [b] | 278 [b] |
| EtOH | 20 | 32.54 [f] | SA | 3 | >100.00 [c] | 14.08 [c] |
| n-C8H17OH | 3 | 1.25 [g] | paracetamol | 9.7 | 42.23 [c] | 10.69[c] |
| MeOH | 33 | 38.46 [h] | ibuprofen | 4.4 | 5.9 [c] | 9.9 [c] |
| CO(CH3)2 | 29 | 33.33 [h] | diclofenac | 3.9 | 0.26 [c] | 0.01 [c] |
| AcOH | 12 | 7.59 [h] | ketoprofen | 3.9 | 0.11 [c] | 0.88 [c] |
| CHCl3 | 6 | 2.22 [h] | piroxicam | 6.3 | 2.68 [c] | 2.11 [c] |
| O(Et)2 | 5 | 32.82 * [h] | celecoxib | 11.1 | 15 [b] | 0.04 [b] |
The bioavailability of most medicinal substances in the lumen of the gastrointestinal tract (GI) is increased when the drug is in a non-ionized state. Only the undissociated molecule is lipophilic enough to pass through the stomach or intestinal wall by passive diffusion. Due to its chemical structure acetylsalicylic acid shows a pH-dependent equilibrium of the ionized (hydrophilic) and the non-ionized (lipophilic) form of the salicylate [50]. ASA is a weak acid that is poorly dissociated in setting the pH of gastric fluid, and therefore rapidly resorbed by various gastric cell membranes [51]. ASA dissociates to a greater extent in the intestine due to the higher pH in this part of the GI, thus improving its solubility [52]. The higher ionized rate determines the better absorption of ASA in the small intestine than in the stomach at the same pH range. At pH 3.5 or 6.5, the intestinal absorption of ASA is greater than the absorption of the compound in the stomach. The stomach does not absorb ASA at a pH of 6.5 (e.g., the gastric pH of infants ranges from 6 to 8).
3.2. Mechanism of Action for ASA
At present, no one doubts that the known mechanisms of action for ASA fall into two categories, they are cyclooxygenase (COX, alternate name Prostaglandin H2 synthase PGHS, EC.1.14.99.1 [53]), i.e., COX-dependent and COX-independent pathways [54]. When ASA is absorbed into the blood, it breaks down into two pharmacologically important molecules: active acetate and salicylate [17]. From a chemical point of view, the primary mechanism of action for ASA is the transfer of the Ac group to -OH and -NH2 functionalities present in biological macromolecules [12]. It has been proven that ASA reacts with nucleophilic groups on proteins (such as Lys and Arg (with -NH2 group), Ser, Thr, Tyr (with -OH group, and Cys (with -SH group)) resulting in irreversible acetylation. From a biological point of view, the key mechanism of action for ASA is the irreversible inhibition of prostaglandin peroxide synthase 1 (COX-1), 2 (COX-2) and 3 (COX-3)) of the prostaglandin precursor and thromboxane producing enzymes [55][56][57]. COX is a membrane enzyme endowed with a long and narrow hydrophobic enzyme tunnel that runs from the surface of the membrane into the interior of the protein molecule [58] (Figure 3). COX-1 is structurally and enzymatically similar to COX-2 [59].
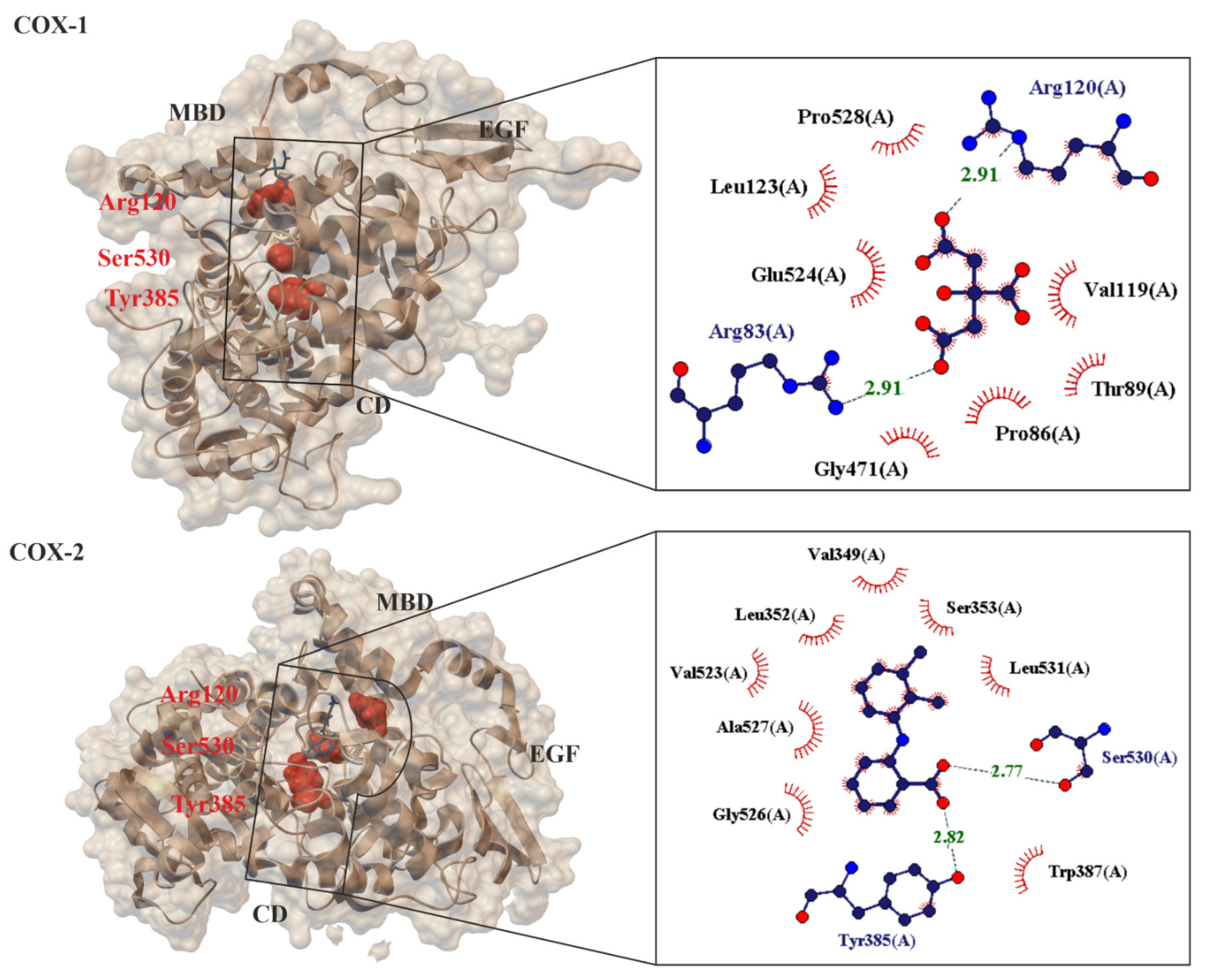
Figure 3. Comparison of 3-D crystal structures of human cyclooxygenase COX-1 (PDB 6y3c, 3.36 Å) [60] and COX-2 (PDB: 5ikr; 2.34 Å) [61] highlighting the bound ligands, important protein domains (such as EGF: epidermal growth factor domain, MBD: membrane binding domain and CD: catalytic domain) and amino acids (Arg120, Ser530, Tyr385). Enlarged area showing structural elements around acidic ligand binding site. Residues forming hydrogen bonds (dashed lines) with the crystalized ligand are shown in spherical form with interatomic distances in Å. Residues forming Van der Waals interactions with ligands are shown as labeled arcs with radial spokes pointing toward the ligand atoms.
The small difference in size between the active sites of COX-1 and COX-2 has been exploited by pharmaceutical companies to develop selective COX-2 inhibitors, such as celecoxib [62], rofecoxib [63] and meloxicam [64][65], which reduce inflammation without damaging the stomach mucosa [66]. COX-3 is a splicing variant of COX-1 which has retained intron-1 during translation and which is found in human tissues in a polyadenylated form. Selective inhibition of COX-3 will discover potent and valuable new drugs for controlling pain and fever [66]. ASA acetylates SER-530 COX-1 thereby block prostaglandins (PGs) and thromboxane A2 (TxA2) synthesis in platelets and reduce platelet aggregation. A vasoconstrictor, TxA2, also aids in the aggregation of platelets during hemostasis (Figure 4). This mechanism of action explains the effect of ASA in preventing thrombosis of the coronary arteries and cerebral vessels [67][68]. These result in a decreased chance of thrombosis or thrombotic events, but the exact role of ASA in primary prevention in still uncertain [56][69].
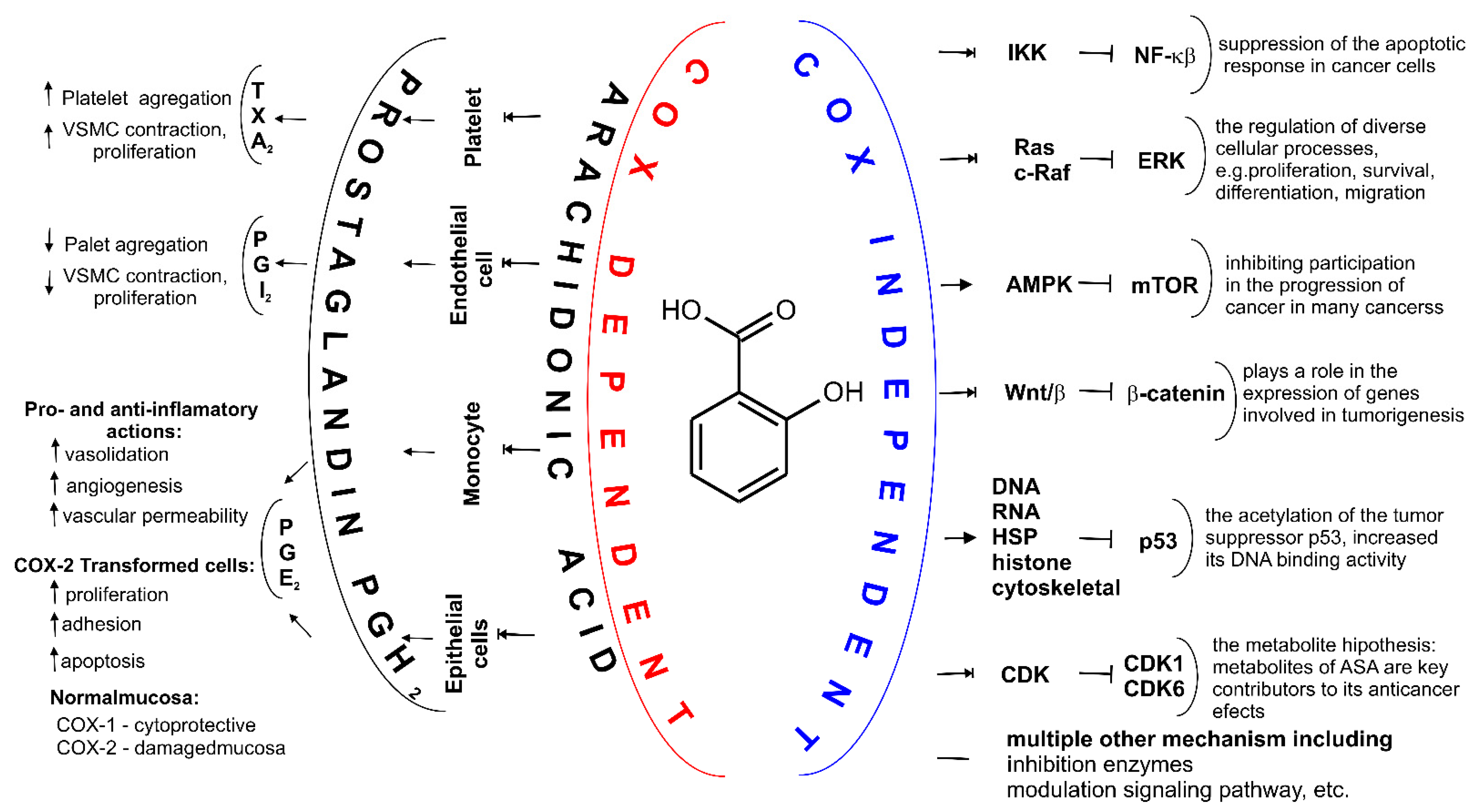
Figure 4. Two major mechanisms of action for ASA. Depending on the cell type COX-dependent mechanism affects synthesis of different prostaglandins (PGs) including PGH2, PGE2, PGF2α, prostacyclin PGI2 and tromboxane A2 (TXA2). Through various COX-independent mechanisms, ASA may modulate carcinogenesis and alter growth rate of the intestinal tumor. These pathways, including inhibition of nuclear factor (NF)-κB signaling and the extracellular signal-regulated kinase (ERK) signaling, cells and may arrest cell growth by inhibiting cyclin dependent kinase CDKs, inhibition of the Wing-like glycoproteins, i.e., Wnt signaling, β-Catenin phosphorylation, activation adenosine monophosphate activated protein kinase (AMPK), decreasing mechanistic target of rapamycin (mTOR) signaling, acetylation many proteins such as histones, cytoskeletal and heat shock proteins (HSP), glycolytic and pentose pathway enzymes, proteasomal subunits and mitochondrial proteins, proteins involved in translation, wild-type and mutant a tumor suppressor protein (p53), and multiple other mechanisms, e.g., inhibition hydroperoxy fatty acid peroxidase in the lipoxygenase pathway of arachidonic acid metabolism.
ASA is the only NSAID that covalently modifies the COX enzymes. ASA binds to SER- 516 residue on the active site of COX-2 in the same fashion as its binding to the active site of COX-1. SER-530 is located near COX-1 active site and the Ac group added to this SER residue hinders the access of substrate (arachidonic acid, AA) to the active site [70]. COX-1 and COX-2 are isoenzymes (60% sequence identity) and their active site structure are similar too [71]. COX-2 is inducible, and its expression is enhanced by the same prostaglandins that are synthesized by COX-1 in platelets and epithelial cells [55]. However, COX-2 has a side pocket in the substrate channel that is not in COX-1. Additionally, the substrate channel of COX-2 is larger and more flexible. These structural differences make ASA (at high doses, i.e., micromolar to millimolar) about 60-fold more potent to inhibit platelet COX-1 than monocyte COX-2 (as measured on prostanoid biosynthesis by washed human platelets (expressing COX-1) and isolated monocytes (expressing COX-2)) [46]), and 170 times more effective at inhibiting COX-1 anti-inflammatory activity of a COX-isozyme activity as IC50 ratio of COX-2/COX-1 [72]. COX-2 gene expression is induced by myriad stimuli and is sustained by a positive feedback mechanism via prostaglandins (PGE2 and PGI2). These prostaglandins activate adenylyl cyclase (GC), thereby increasing intracellular cyclic AMP levels and enhancing COX-2 transcription via the protein kinase cAMP-dependent (PKA) pathway [73] (Figure 4). A third cyclooxygenase, COX-3, is selectively inhibited by low concentrations of some NSAID including ASA. Blocking or changing COX activities are the result of acetylation by the active acetate released from the ASA molecule and not by salicylate [74]. This has been shown in platelet studies, where a low concentration of ASA is sufficient to irreversibly block COX-1, while salicylate is inactive. In humans, deacetylation is so intense in the intestinal lumen, portal circulation and liver that it is assumed that only about 50% of the ASA dose reaches the systemic circulation unchanged [39][75]. Low-dose ASA provides a paradigm of COX isozyme-selective and cell-specific inhibition, by virtue of its short half-life and its ability to inactivate COX irreversibly. Other NSAIDs lack these unique pharmacokinetic and pharmacodynamic features and do not usually achieve the same degree of persistent platelet COX-1 inhibition as is obtained with low-dose ASA [76].
However, other COX independent ASA mechanisms have gradually been identified. Subsequent studies aimed to identify additional effects of this drug (such as chemopreventive or other) that could help explain its pharmacological properties. ASA can modulate many different signaling pathways. It was found to inhibit activated B cell nuclear factor κ-light chain enhancer (NF-kB) without affecting other transcription factors. It was first demonstrated in 1994 [77]. ASA inhibits the activation of NF-kB, thus prevents the degradation of the NF-kB inhibitor, I kappa B, and therefore is retained in the cytosol. ASA also inhibits NF-kB in B-dependent transcription from the Ig kappa (Igκ) enhancer and the human immunodeficiency virus (HIV) long terminal repeat (LTR) in transfected T cells [77]. It has also been demonstrated that ASA alone is capable of stimulating hippocampal plasticity. It was observed that ASA binds to PPARα at the Tyr314 residue of its ligand-binding domain (LBD).
On binding to the PPARα LBD, ASA induces activation of PPARα to upregulate transcription of the cyclic adenosine monophosphate response element-binding protein (CREB) and associated hippocampal plasticity and associated hippocampal plasticity [78]. Furthermore, low-dose of ASA treatment increased the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-and N-methyl-D-aspartate (NMDA)-mediated calcium current in hippocampal slices and improved memory and learning in the FAD5X, but not FAD5X/Ppara-null, mice [78]. However, analgesic doses of ASA do not cause psychiatric disturbances such as hypnosis or rapid mood changes, relieve pain without affecting other sensory modalities and do not modify excitatory mechanisms involving the brainstem reticular formation. Therefore, it seems very likely that any central mechanism is probably subcortical, perhaps at the level of the hypothalamus. The small number of such effects in the CNS also suggests that the analgesic action of salicylates is largely through their peripheral action [79].
So far, many mechanisms have been proposed in which it has been proven that ASA influences transcription factors [54][80][81][82], cell signaling [83][84] and mitochondrial function [85], as well as the activity of various enzymes by many different action types (Figure 4, Table 2) [86][87]. ASA induces autophagy and stimulates mitophagy. However, not all of the exact mechanisms by which ASA works are not still known and understood.
Table 2. Compilation of different therapeutic targets and their action type for ASA [2][3][70][88][89][90][91][92].
| Drug Targets Name | Action Type |
|---|---|
| Prostaglandin G/H synthase 1, Prostaglandin G/H synthase 2, Aldo-keto reductase family 1 member C1, Endothelin-1 receptor, Ribosomal protein S6 kinase alpha-3, NF-kappa-B inhibitor alpha, tumor necrosis factor-inducible gene 6 protein, Caspase-1, Caspase-3, Solute carrier family 22 member 6, Solute carrier family 22 member 8 | Inhibitor |
| 5′-AMP-activated protein kinase, | Activator |
| Cellular tumor antigen p53, Cytochrome P450 2C19, P-glycoprotein 1, | Inducer |
| Cytochrome P450 2C9, UDP-glucuronosyltransferase 1–6, Arylamine N-acetyltransferase 2, P-glycoprotein 1 | Substrate |
| 78 kDa glucose-regulated protein, | Binding |
| P-glycoprotein 1 | Modulator |
| Tumor necrosis factor-inducible gene 6 protein, Caspase-1, Caspase-3, G1/S-specific cyclin-D1, Myc proto-oncogene protein, Proliferating cell nuclear antigen, Cyclin A, | Downregulator |
This entry is adapted from the peer-reviewed paper 10.3390/molecules27238412
References
- Anatomical Therapeutic Chemical Classification System. Available online: https://www.whocc.no/atc_ddd_index/?code=N02BA01 (accessed on 10 October 2022).
- DrugBank. Available online: https://go.drugbank.com/drugs/DB00945 (accessed on 10 October 2022).
- Drug Index A to Z. Available online: https://www.drugs.com/search.php?searchterm=aspirin&sources%5B%5D= (accessed on 10 October 2022).
- Balescu, A.; Nedelcu, L. The aspirin-the first drug obtained by sinthesys-frequently used currently. Bull. Transilv. Univ. Brasov. Med. Sci. Ser. VI 2009, 6, 145.
- Hybiak, J.; Broniarek, I.; Kiryczyński, G.; Los, L.D.; Rosik, J.; Machaj, F.; Sławiński, H.; Jankowska, K.; Urasińska, E. Aspirin and its pleiotropic application. Eur. J. Pharmacol. 2020, 866, 172762.
- Marko, V. From Aspirin to Viagra: Stories of the Drugs That Changed the World; Springer Nature: Berlin, Germany, 2020.
- Warner, T.D.; Mitchell, J.A. Cyclooxygenase-3 (COX-3): Filling in the gaps toward a COX continuum? Proc. Natl. Acad. Sci. USA 2002, 99, 13371–13373.
- Zdrojewicz, Z.; Jagodziński, A.; Kowalik-Jagodzińska, M.; Zielińska, E. Stare i nowe oblicze aspiryny. Pediatr. Med. Rodz. 2018, 14, 369–375.
- Wood, J.N. From plant extract to molecular panacea: A commentary on Stone (1763) ‘An account of the success of the bark of the willow in the cure of the agues’. Philos. Transac. R. Soc. B Biol. Sci. 2015, 370, 20140317.
- Jack, D.B. One hundred years of aspirin. Lancet 1997, 350, 437–439.
- Rocke, A.J. Hermann Kolbe, Encyclopedia Britannica. Available online: https://www.britannica.com/biography/Hermann-Kolbe (accessed on 10 October 2022).
- Rocke, A.J. The Quiet Revolution: Hermann Kolbe and the Science of Organic Chemistry; University of California Press: Berkeley, CA, USA, 1993.
- Alegbeleye, B.J.; Akpoveso, O.O.P.; Mohammed, R.K.; Asare, B.Y.A. Pharmacology, Pharmaceutics and Clinical Use of Aspirin: A Narrative Review. J. Drug Deliv. Ther. 2020, 10, 236–253.
- Gulyás, A.; Heszberger, Z.; Biró, J. Amazing Scientific Discoveries: Aspirin, Cattle, Business Communication and Others. In Paths; Springer: Berlin, Germany, 2021; pp. 67–71.
- Montinari, M.R.; Minelli, S.; De Caterina, R. The first 3500 years of aspirin history from its roots—A concise summary. Vasc. Pharmacol. 2019, 113, 1–8.
- Leggat, P.A. A brief history of the discovery and applications of aspirin. Ann. ACTM 2012, 13, 5–9.
- Desborough, M.J.; Keeling, D.M. The aspirin story–from willow to wonder drug. Br. J. Haematol. 2017, 177, 674–683.
- Wick, J. Aspirin: A history, a love story. Consult. Pharm. 2012, 27, 322–329.
- Nowaczyk, A. 120 urodziny aspiryny. Wiad. Akad. 2019, 76, 21–25.
- Nowaczyk, A. Fenomen aspiryny. Głos Uczelni 2019, XI–XII, 39–44.
- The U.S. Food and Drug Administration (FDA). Clinical Trials Register. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=aspirin&cntry=&state=&city=&dist= (accessed on 1 October 2022).
- The European Union. Clinical Trials Register. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=aspirin (accessed on 1 October 2022).
- The Australian New Zealand Clinical Trials Registry. Available online: https://www.australianclinicaltrials.gov.au/anzctr_feed/form (accessed on 1 October 2022).
- Saleh, I. The Pattern of Aspirin Use in Patients with Heart Failure at Mohammad Hoesin Hospital Palembang from 1st June 2013–30th July 2014. Int. J. Health Sci. Res. 2016, 13–24.
- The Cambridge Structural Database (CSD). Available online: https://www.ccdc.cam.ac.uk/structures/? (accessed on 10 October 2022).
- Wheatley, P. 1163. The crystal and molecular structure of aspirin. J. Chem. Soc. (Resumed) 1964, 6036–6048.
- Kim, Y.; Machida, K.; Taga, T.; Osaki, K. Structure redetermination and packing analysis of aspirin crystal. Chem. Pharm. Bull. 1985, 33, 2641–2647.
- Ouvrard, C.; Price, S.L. Toward crystal structure prediction for conformationally flexible molecules: The headaches illustrated by aspirin. Cryst. Growth Des. 2004, 4, 1119–1127.
- Payne, R.; Rowe, R.C.; Roberts, R.; Charlton, M.; Docherty, R. Potential polymorphs of aspirin. J. Comput. Chem. 1999, 20, 262–273.
- Bond, A.D.; Solanko, K.A.; Parsons, S.; Redder, S.; Boese, R. Single crystals of aspirin form II: Crystallisation and stability. Cryst. Eng. Comm. 2011, 13, 399–401.
- Bernstein, J. Polymorphism in Molecular Crystals 2e; International Union of Crystal: Chester UK; Oxford University Press: Oxford, UK, 2020; Volume 30.
- FDA Guidance for Industry: Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q6a-specifications-test-procedures-and-acceptance-criteria-new-drug-substances-and-new-drug-products (accessed on 1 October 2022).
- Tsuri, Y.; Maruyama, M.; Fujimoto, R.; Okada, S.; Adachi, H.; Yoshikawa, H.Y.; Takano, K.; Murakami, S.; Matsumura, H.; Inoue, T. Crystallization of aspirin form II by femtosecond laser irradiation. Appl. Phys. Express 2018, 12, 015507.
- Yadav, A.R.; Mohite, S.K. Different techniques and characterization of polymorphism with their evaluation: A Review. Asian J. Pharm. Technol. 2020, 10, 213–216.
- Vishweshwar, P.; McMahon, J.A.; Oliveira, M.; Peterson, M.L.; Zaworotko, M.J. The predictably elusive form II of aspirin. J. Am. Chem. Soc. 2005, 127, 16802–16803.
- Crowell, E.L.; Dreger, Z.A.; Gupta, Y.M. High-pressure polymorphism of acetylsalicylic acid (aspirin): Raman spectroscopy. J. Mol. Struct. 2015, 1082, 29–37.
- Shtukenberg, A.G.; Hu, C.T.; Zhu, Q.; Schmidt, M.U.; Xu, W.; Tan, M.; Kahr, B. The third ambient aspirin polymorph. Cryst. Growth Des. 2017, 17, 3562–3566.
- Choudhary, A.; Kamer, K.J.; Raines, R.T. An n→ π* interaction in aspirin: Implications for structure and reactivity. J. Organ. Chem. 2011, 76, 7933–7937.
- Lee, E.H. A practical guide to pharmaceutical polymorph screening & selection. Asian J. Pharm. Sci. 2014, 9, 163–175.
- Dymczuk, K. Study of Phase Equilibria in Binary System: Pharmaceuticals + Solvent; Diss. Zakład Chemii Fizycznej, Politechnika Warszwska: Warsaw, Poland, 2012.
- Poźniak, B.; Świtała, M. Salicylany w farmakoterapii weterynaryjnej. Część I. Współczesny stan wiedzy o właściwościach farmakologicznych. Życie Weter 2012, 87, 942–947.
- Perlovich, G.L.; Bauer-Brandl, A. Thermodynamics of solutions I: Benzoic acid and acetylsalicylic acid as models for drug substances and the prediction of solubility. Pharm. Res. 2003, 20, 471–478.
- Gaohua, L.; Miao, X.; Dou, L. Crosstalk of physiological pH and chemical pKa under the umbrella of physiologically based pharmacokinetic modeling of drug absorption, distribution, metabolism, excretion, and toxicity. Expert Opin. Drug Met. 2021, 17, 1103–1124.
- Nordström, F.L.; Rasmuson, Å.C. Solubility and melting properties of salicylic acid. J. Chem. Eng. Data 2006, 51, 1668–1671.
- Chen, C.-C.; Song, Y. Solubility modeling with a nonrandom two-liquid segment activity coefficient model. Ind. Eng. Chem. Res. 2004, 43, 8354–8362.
- Vane, J.; Bakhle, Y.; Botting, R. Cyclooxygenase 1 and 2. Annu. Rev. Pharmacol. 1998, 38, 97–120.
- Cryer, B.; Feldman, M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 413–421.
- Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013, 191, 39–65.
- Nokhodchi, A.; Ghafourian, T.; Nashed, N.; Asare-Addo, K.; Behboudi, E.; Sefid-Sefidehkhan, Y.; Zarghampour, A.; Rahimpour, E.; Jouyban, A. Solubility Study of Acetylsalicylic Acid in Ethanol+ Water Mixtures: Measurement, Mathematical Modeling, and Stability Discussion. AAPS Pharm. Sci. Tech. 2022, 23, 1–12.
- Shalmashi, A.; Eliassi, A. Solubility of salicylic acid in water, ethanol, carbon tetrachloride, ethyl acetate, and xylene. J. Chem. Eng. Data 2008, 53, 199–200.
- Belhachemi, B.; Makhlouf, H.; Belhachemi, M.H. Determination and Correlation of Solubilities of Benzoic Acid, Salicylic Acid, Resorcinol and Hydroquinone in Water and in 1-Octanol at Temperatures from 297.25 K to 334.45 K. Int. J. Thermophys. 2021, 42, 1–16.
- Stevens, H.; Voelker, M.; Gow, L.; MacDougall, F.; Bieri, G. In-vivo disintegration and absorption of two fast acting aspirin tablet formulations compared to ibuprofen tablets using pharmacoscintigraphy. J. Drug Deliv. mSci. Technol. 2019, 51, 535–541.
- Jirmář, R.; Widimský, P. Enteric-coated aspirin in cardiac patients: Is it less effective than plain aspirin? Cor et Vasa 2018, 60, e165–e168.
- Kanani, K.; Gatoulis, S.C.; Voelker, M. Influence of differing analgesic formulations of aspirin on pharmacokinetic parameters. Pharmaceutics 2015, 7, 188–198.
- Brenda The Compensive Enzyme Information Sysytem. Available online: https://www.brenda-enzymes.org/enzyme.php?ecno=1.14.99.1 (accessed on 10 October 2022).
- Sankaranarayanan, R.; Kumar, D.R.; Altinoz, M.A.; Bhat, G.J. Mechanisms of colorectal cancer prevention by aspirin—a literature review and perspective on the role of COX-dependent and-independent pathways. Int. J. Mol. Sci. 2020, 21, 9018.
- Zacharias-Millward, N.; Menter, D.G.; Davis, J.S.; Lichtenberger, L.; Hawke, D.; Hawk, E.; Vilar, E.; Bhattacharya, P.; Millward, S. Beyond COX-1: The effects of aspirin on platelet biology and potential mechanisms of chemoprevention. Cancer Metast. Rev. 2017, 36, 289–303.
- Fuster, V.; Sweeny, J.M. Aspirin: A historical and contemporary therapeutic overview. Circulation 2011, 123, 768–778.
- Gupta, K.; Selinsky, B.S.; Kaub, C.J.; Katz, A.K.; Loll, P.J. The 2.0 Å resolution crystal structure of prostaglandin H2 synthase-1: Structural insights into an unusual peroxidase. J. Mol. Biol. 2004, 335, 503–518.
- Garavito, R.M.; Malkowski, M.G.; DeWitt, D.L. The structures of prostaglandin endoperoxide H synthases-1 and-2. Prostaglandins Other Lipid Mediat. 2002, 68, 129–152.
- Miciaccia, M.; Belviso, B.D.; Iaselli, M.; Cingolani, G.; Ferorelli, S.; Cappellari, M.; Loguercio Polosa, P.; Perrone, M.G.; Caliandro, R.; Scilimati, A. Three-dimensional structure of human cyclooxygenase (hCOX)-1. Sci. Rep. 2021, 11, 4312.
- Orlando, B.J.; Malkowski, M.G. Substrate-selective inhibition of cyclooxygeanse-2 by fenamic acid derivatives is dependent on peroxide tone. J. Biol. Chem. 2016, 291, 15069–15081.
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. JAMA 2000, 284, 1247–1255.
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Schnitzer, T.J. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N. Engl. J. Med. 2000, 343, 1520–1528.
- Dequeker, J.; Hawkey, C.; Kahan, A.O.; Steinbrück, K.; Alegre, C.; Baumelou, E.; Begaud, B.; Isomäki, H.; Littlejohn, G.; Mau, J. Improvement in gastrointestinal tolerability of the selective cyclooxygenase (COX)-2 inhibitor, meloxicam, compared with piroxicam: Results of the Safety and Efficacy Large-scale Evaluation of COX-inhibiting Therapies (SELECT) trial in osteoarthritis. Br. J. Rheumatol. 1998, 37, 946–951.
- Nowaczyk, A.; Szwedowski, D.; Dallo, I.; Nowaczyk, J. Overview of First-Line and Second-Line Pharmacotherapies for Osteoarthritis with Special Focus on Intra-Articular Treatment. IJMS 2022, 23, 1566.
- Vane, J.; Botting, R. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258.
- Wu, K.K. Aspirin and Other Cyclooxygenase Inhibitors: New Therapeutic Insights; Seminars in vascular medicine; Thieme Medical Publishers, Inc.: New York, NY, USA, 2003; pp. 107–112.
- Bianconi, V.; Violi, F.; Fallarino, F.; Pignatelli, P.; Sahebkar, A.; Pirro, M. Is acetylsalicylic acid a safe and potentially useful choice for adult patients with COVID-19? Drugs 2020, 80, 1383–1396.
- Stolarek, W.; Kasprzak, M.; Sikora, J.; Siemińska, E.; Grześk, G. High on-treatment platelet reactivity to aspirin in patients after myocardial infarction. Biomed. Pharmacother. 2022, 147, 112618.
- Original Aspirin. The Summary of Product Characteristic. pp. 129–152. Available online: https://www.medicines.org.uk/emc/product/13691/pil (accessed on 1 October 2022).
- Zarghi, A.; Arfaei, S. Selective COX-2 inhibitors: A review of their structure-activity relationships. IJPR 2011, 10, 655.
- Altman, R.; Luciardi, H.L.; Muntaner, J.; Herrera, R.N. The antithrombotic profile of aspirin. Aspirin resistance, or simply failure? Thromb. J. 2004, 2, 1–8.
- Patrono, C. The multifaceted clinical readouts of platelet inhibition by low-dose aspirin. J. Am. Coll. Cardiol. 2015, 66, 74–85.
- Meade, E.A.; Smith, W.L.; Dewitt, D.L. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1993, 268, 6610–6614.
- Costello, P.B.; Green, F.A. Identification and partial purification of the major aspirin hydrolyzing enzyme in human blood. Arthritis Rheum. 1983, 26, 541–547.
- Patrono, C.; Patrignani, P.; Rodríguez, L.A.G. Cyclooxygenase-selective inhibition of prostanoid formation: Transducing biochemical selectivity into clinical read-outs. J. Clin. Investig. 2001, 108, 7–13.
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959.
- Patel, D.; Roy, A.; Kundu, M.; Jana, M.; Luan, C.-H.; Gonzalez, F.J.; Pahan, K. Aspirin binds to PPARα to stimulate hippocampal plasticity and protect memory. Proc. Natl. Acad. Sci. USA 2018, 115, E7408–E7417.
- Clissold, S.P. Aspirin and related derivatives of salicylic acid. Drugs 1986, 32, 8–26.
- Shishodia, S.; Aggarwal, B.B. Nuclear factor-κB activation: A question of life or death. BMB Rep. 2002, 35, 28–40.
- Kutuk, O.; Basaga, H. Aspirin inhibits TNFα-and IL-1-induced NF-κB activation and sensitizes HeLa cells to apoptosis. Cytokine 2004, 25, 229–237.
- Tran, P.H.; Lee, B.-J.; Tran, T.T. Current studies of aspirin as an anticancer agent and strategies to strengthen its therapeutic application in cancer. Curr. Pharm. Des. 2021, 27, 2209–2220.
- Kim, J.; Lee, K.-S.; Kim, J.-H.; Lee, D.-K.; Park, M.; Choi, S.; Park, W.; Kim, S.; Choi, Y.K.; Hwang, J.Y. Aspirin prevents TNF-α-induced endothelial cell dysfunction by regulating the NF-κB-dependent miR-155/eNOS pathway: Role of a miR-155/eNOS axis in preeclampsia. Free Radic. Biol. Med. 2017, 104, 185–198.
- Li, X.; Wu, S.; Yu, Y. Aspirin Use and the Incidence of Hepatocellular Carcinoma in Patients with Hepatitis B Virus or Hepatitis C Virus Infection: A Meta-Analysis of Cohort Studies. Front. Med. 2021, 7, 968.
- Zhang, H.; Lu, J.; Jiao, Y.; Chen, Q.; Li, M.; Wang, Z.; Yu, Z.; Huang, X.; Yao, A.; Gao, Q. Aspirin inhibits natural killer/T-cell lymphoma by modulation of VEGF expression and mitochondrial function. Front. Oncol. 2019, 8, 679.
- Joshi, S.; Murphy, E.; Olaniyi, P.; Bryant, R. The multiple effects of aspirin in prostate cancer patients. Cancer Treat. Res. Commun. 2021, 26, 100267.
- Patrono, C.; Baigent, C. Role of aspirin in primary prevention of cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 675–686.
- RCSB Database. Available online: https://www.rcsb.org/ligand/AIN (accessed on 1 October 2022).
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Revi. Drug Discov. 2004, 3, 673–683.
- Oprea, T.; Mestres, J. Drug repurposing: Far beyond new targets for old drugs. AAPS J. 2012, 14, 759–763.
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.-H.D. Drug repositioning and repurposing: Terminology and definitions in literature. Drug Discov. Today 2015, 20, 1027–1034.
This entry is offline, you can click here to edit this entry!