Pancreatic ductal adenocarcinoma (PDAC) has an extremely poor prognosis due to the lack of methods or biomarkers for early diagnosis and its resistance to conventional treatment modalities, targeted therapies, and immunotherapies. PDACs are a heterogenous group of malignant epithelial neoplasms with various histomorphological patterns and complex, heterogenous genetic/molecular landscapes. The newly proposed molecular classifications of PDAC based on extensive genomic, transcriptomic, proteomic and epigenetic data have provided significant insights into the molecular heterogeneity and aggressive biology of this deadly disease. Studies characterizing the tumor microenvironment (TME) have shed light on the dynamic interplays between the tumor cells and the immunosuppressive TME of PDAC, which is essential to disease progression, as well as its resistance to chemotherapy, newly developed targeted therapy and immunotherapy. There is a critical need for the development of predictive markers that can be clinically utilized to select effective personalized therapies for PDAC patients.
- pancreatic ductal adenocarcinoma
- molecular pathology
- predictive marker
- tumor microenvironment
- targeted therapy and immunotherapy
1. Introduction
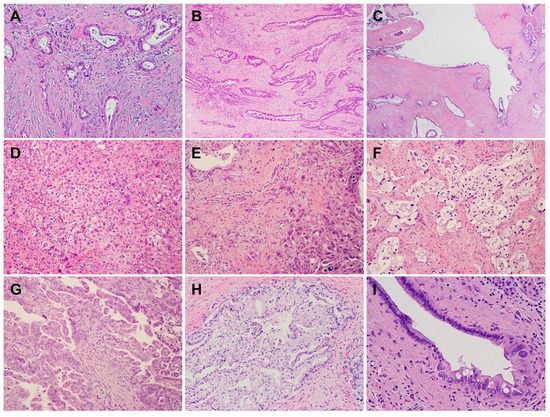
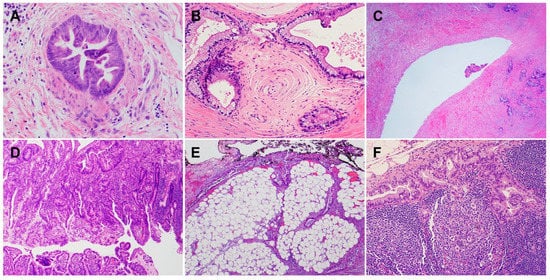
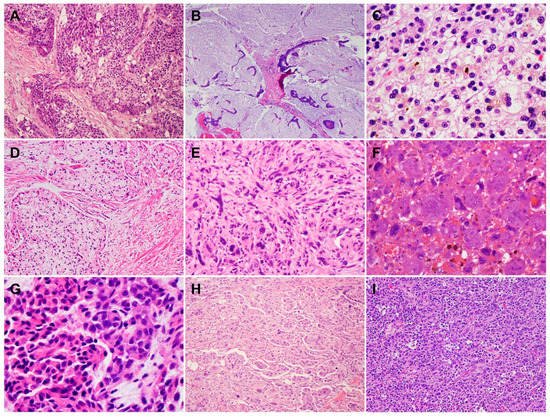
2. Histology and Morphologic Heterogeneity of PDAC
3. Genetic Alterations and Molecular Subtypes of PDAC
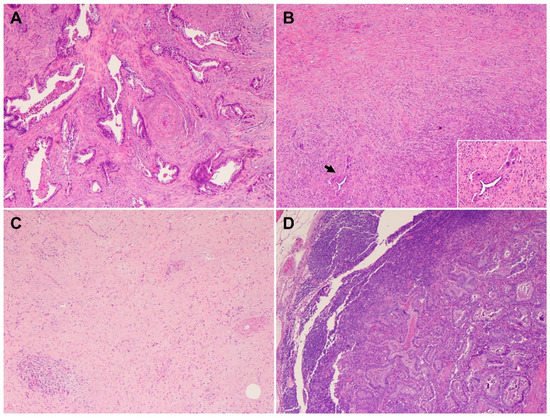
4. Heterogeneous Response of PDAC to Neoadjuvant Therapy
This entry is adapted from the peer-reviewed paper 10.3390/cells11193068
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708.
- National Cancer Institute SRP, Cancer Statistics Branch. Surveillance Epidemiology and End Results (SEER) (1975–2018). 2021. Available online: http://seer.cancer.gov (accessed on 7 August 2022).
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85.
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188.
- Garcea, G.; Dennison, A.R.; Pattenden, C.J.; Neal, C.P.; Sutton, C.D.; Berry, D.P. Survival following curative resection for pancreatic ductal adenocarcinoma. A systematic review of the literature. JOP 2008, 9, 99–132.
- Hong, S.M.; Park, J.Y.; Hruban, R.H.; Goggins, M. Molecular signatures of pancreatic cancer. Arch. Pathol. Lab. Med. 2011, 135, 716–727.
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113.
- Chatterjee, D.; Katz, M.H.; Rashid, A.; Wang, H.; Iuga, A.C.; Varadhachary, G.R.; Wolff, R.A.; Lee, J.E.; Pisters, P.W.; Crane, C.H.; et al. Perineural and intraneural invasion in posttherapy pancreaticoduodenectomy specimens predicts poor prognosis in patients with pancreatic ductal adenocarcinoma. Am. J. Surg. Pathol. 2012, 36, 409–417.
- Chatterjee, D.; Rashid, A.; Wang, H.; Katz, M.H.; Wolff, R.A.; Varadhachary, G.R.; Lee, J.E.; Pisters, P.W.; Gomez, H.F.; Abbruzzese, J.L.; et al. Tumor invasion of muscular vessels predicts poor prognosis in patients with pancreatic ductal adenocarcinoma who have received neoadjuvant therapy and pancreaticoduodenectomy. Am. J. Surg. Pathol. 2012, 36, 552–559.
- Fischer, L.K.; Katz, M.H.; Lee, S.M.; Liu, L.; Wang, H.; Varadhachary, G.R.; Wolff, R.A.; Lee, J.E.; Maitra, A.; Roland, C.L.; et al. The number and ratio of positive lymph nodes affect pancreatic cancer patient survival after neoadjuvant therapy and pancreaticoduodenectomy. Histopathology 2016, 68, 210–220.
- Liu, L.; Katz, M.H.; Lee, S.M.; Fischer, L.K.; Prakash, L.; Parker, N.; Wang, H.; Varadhachary, G.R.; Wolff, R.A.; Lee, J.E.; et al. Superior Mesenteric Artery Margin of Posttherapy Pancreaticoduodenectomy and Prognosis in Patients with Pancreatic Ductal Adenocarcinoma. Am. J. Surg. Pathol. 2015, 39, 1395–1403.
- International Agency for Research on Cancer. WHO 2019 Classification of Tumors, Digestive System Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2019; Volume 1, pp. 295–332.
- Adsay, N.V.; Merati, K.; Nassar, H.; Shia, J.; Sarkar, F.; Pierson, C.R.; Cheng, J.D.; Visscher, D.W.; Hruban, R.H.; Klimstra, D.S. Pathogenesis of colloid (pure mucinous) carcinoma of exocrine organs: Coupling of gel-forming mucin (MUC2) production with altered cell polarity and abnormal cell-stroma interaction may be the key factor in the morphogenesis and indolent behavior of colloid carcinoma in the breast and pancreas. Am. J. Surg. Pathol. 2003, 27, 571–578.
- Adsay, N.V.; Pierson, C.; Sarkar, F.; Abrams, J.; Weaver, D.; Conlon, K.C.; Brennan, M.F.; Klimstra, D.S. Colloid (mucinous noncystic) carcinoma of the pancreas. Am. J. Surg. Pathol. 2001, 25, 26–42.
- Poultsides, G.A.; Reddy, S.; Cameron, J.L.; Hruban, R.H.; Pawlik, T.M.; Ahuja, N.; Jain, A.; Edil, B.H.; Iacobuzio-Donahue, C.A.; Schulick, R.D.; et al. Histopathologic basis for the favorable survival after resection of intraductal papillary mucinous neoplasm-associated invasive adenocarcinoma of the pancreas. Ann. Surg. 2010, 251, 470–476.
- Seidel, G.; Zahurak, M.; Iacobuzio-Donahue, C.; Sohn, T.A.; Adsay, N.V.; Yeo, C.J.; Lillemoe, K.D.; Cameron, J.L.; Hruban, R.H.; Wilentz, R.E. Almost all infiltrating colloid carcinomas of the pancreas and periampullary region arise from in situ papillary neoplasms: A study of 39 cases. Am. J. Surg. Pathol. 2002, 26, 56–63.
- Brody, J.R.; Costantino, C.L.; Potoczek, M.; Cozzitorto, J.; McCue, P.; Yeo, C.J.; Hruban, R.H.; Witkiewicz, A.K. Adenosquamous carcinoma of the pancreas harbors KRAS2, DPC4 and TP53 molecular alterations similar to pancreatic ductal adenocarcinoma. Mod. Pathol. 2009, 22, 651–659.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Wilentz, R.E.; Goggins, M.; Redston, M.; Marcus, V.A.; Adsay, N.V.; Sohn, T.A.; Kadkol, S.S.; Yeo, C.J.; Choti, M.; Zahurak, M.; et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: A newly described and characterized entity. Am. J. Pathol. 2000, 156, 1641–1651.
- Calhoun, E.S.; Jones, J.B.; Ashfaq, R.; Adsay, V.; Baker, S.J.; Valentine, V.; Hempen, P.M.; Hilgers, W.; Yeo, C.J.; Hruban, R.H.; et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: Potential therapeutic targets. Am. J. Pathol. 2003, 163, 1255–1260.
- Goggins, M.; Offerhaus, G.J.; Hilgers, W.; Griffin, C.A.; Shekher, M.; Tang, D.; Sohn, T.A.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am. J. Pathol. 1998, 152, 1501–1507.
- Yamamoto, H.; Itoh, F.; Nakamura, H.; Fukushima, H.; Sasaki, S.; Perucho, M.; Imai, K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001, 61, 3139–3144.
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806.
- Mehlen, P.; Delloye-Bourgeois, C.; Chedotal, A. Novel roles for Slits and netrins: Axon guidance cues as anticancer targets? Nat. Rev. Cancer 2011, 11, 188–197.
- Sabatier, C.; Plump, A.S.; Le, M.; Brose, K.; Tamada, A.; Murakami, F.; Lee, E.Y.; Tessier-Lavigne, M. The divergent Robo family protein rig-1/Robo3 is a negative regulator of slit responsiveness required for midline crossing by commissural axons. Cell 2004, 117, 157–169.
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphorin receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300.
- Samuel, N.; Hudson, T.J. The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 77–87.
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405.
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures with Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783.
- Lowery, M.A.; Wong, W.; Jordan, E.J.; Lee, J.W.; Kemel, Y.; Vijai, J.; Mandelker, D.; Zehir, A.; Capanu, M.; Salo-Mullen, E.; et al. Prospective Evaluation of Germline Alterations in Patients With Exocrine Pancreatic Neoplasms. J. Natl. Cancer Inst. 2018, 110, 1067–1074.
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501.
- Perkhofer, L.; Golan, T.; Cuyle, P.J.; Matysiak-Budnik, T.; Van Laethem, J.L.; Macarulla, T.; Cauchin, E.; Kleger, A.; Beutel, A.K.; Gout, J.; et al. Targeting DNA Damage Repair Mechanisms in Pancreas Cancer. Cancers 2021, 13, 4259.
- Sahin, I.H.; Lowery, M.A.; Stadler, Z.K.; Salo-Mullen, E.; Iacobuzio-Donahue, C.A.; Kelsen, D.P.; O’Reilly, E.M. Genomic instability in pancreatic adenocarcinoma: A new step towards precision medicine and novel therapeutic approaches. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 893–905.
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253 e2244.
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52.
- Cancer Genome Atlas Research Network. Electronic address, a.a.d.h.e.; Cancer Genome Atlas Research, N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203 e113.
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220.
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178.
- Cohen, S.J.; Alpaugh, R.K.; Palazzo, I.; Meropol, N.J.; Rogatko, A.; Xu, Z.; Hoffman, J.P.; Weiner, L.M.; Cheng, J.D. Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 2008, 37, 154–158.
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013 e1993.
- Torres, C.; Grippo, P.J. Pancreatic cancer subtypes: A roadmap for precision medicine. Ann. Med. 2018, 50, 277–287.
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240.
- Hwang, W.L.; Jagadeesh, K.A.; Guo, J.A.; Hoffman, H.I.; Yadollahpour, P.; Reeves, J.W.; Mohan, R.; Drokhlyansky, E.; Van Wittenberghe, N.; Ashenberg, O.; et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat. Genet. 2022, 54, 1178–1191.
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Berson III, A.B.; Cardin, D.B.; Cha, C.; Chiorean, E.G.; Chung, V.; Czito, B.; et al. NCCN Clinical Practice Guidelines in Oncology, Pancreatic Adenocarcinoma (Version 1.2021). Available online: https://www.nccn.org/professionals/physician_gls/PDF/pancreatic.pdf (accessed on 25 January 2021).
- Chatterjee, D.; Katz, M.H.; Rashid, A.; Varadhachary, G.R.; Wolff, R.A.; Wang, H.; Lee, J.E.; Pisters, P.W.; Vauthey, J.N.; Crane, C.; et al. Histologic grading of the extent of residual carcinoma following neoadjuvant chemoradiation in pancreatic ductal adenocarcinoma: A predictor for patient outcome. Cancer 2012, 118, 3182–3190.
- Chou, A.; Ahadi, M.; Arena, J.; Sioson, L.; Sheen, A.; Fuchs, T.L.; Pavlakis, N.; Clarke, S.; Kneebone, A.; Hruby, G.; et al. A Critical Assessment of Postneoadjuvant Therapy Pancreatic Cancer Regression Grading Schemes with a Proposal for a Novel Approach. Am. J. Surg. Pathol. 2021, 45, 394–404.
- Lee, S.M.; Katz, M.H.; Liu, L.; Sundar, M.; Wang, H.; Varadhachary, G.R.; Wolff, R.A.; Lee, J.E.; Maitra, A.; Fleming, J.B.; et al. Validation of a Proposed Tumor Regression Grading Scheme for Pancreatic Ductal Adenocarcinoma After Neoadjuvant Therapy as a Prognostic Indicator for Survival. Am. J. Surg. Pathol. 2016, 40, 1653–1660.
- Nagaria, T.S.; Wang, H.; Chatterjee, D.; Wang, H. Pathology of Treated Pancreatic Ductal Adenocarcinoma and Its Clinical Implications. Arch. Pathol. Lab. Med. 2020, 144, 838–845.