Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Nitric oxide (NO) is implicated in numerous physiological processes, including vascular homeostasis. Reduced NO bioavailability is a hallmark of endothelial dysfunction, a prequel to many cardiovascular diseases. Biomarkers of an early NO-dependent endothelial dysfunction obtained from routine venous blood sampling would be of great interest but are currently lacking.
- nitric oxide
- NO-dependent endothelial function
- NO bioavailability
1. Introduction
The vascular endothelium is one of the largest organs in the body and consists of an active cell monolayer producing a wide range of vascular homeostatic mediators. Nitric Oxide (NO) is the main endothelium-derived substance that maintains the vasculature in a quiescent state by inhibition of contractile tone, cellular proliferation, and thrombosis.
NO was firstly discovered by Joseph Priestley in 1772 [1] as a gas denominated “nitrous air”, but it was not until 1979 that Gruetter highlighted the vasodilator function of NO by demonstrating that precontracted bovine coronary arteries relaxed after exposure to a gas mixture of NO [2]. Few years later, Furchgott found that the ability of rabbit thoracic aorta to relax after exposure to acetylcholine was dependent on the presence of an intact vessel intima, concluding that relaxation involved endothelial cells [3] releasing an endothelium-derived relaxation factor (EDRF). Two independents studies [4,5] in 1987 finally identified NO as this EDRF involved in vascular smooth muscle relaxation through the activation of soluble guanylyl cyclase (sGC). The significance of NO-cGMP was recognized by the 1998 Nobel Prize in Physiology and Medicine awarded to Professors Furchgott, Ignarro and Murad for their discoveries on NO as a signaling molecule in the cardiovascular system [6].
Today, the most commonly accepted functional feature of endothelial dysfunction refers to abnormalities in the regulation of the vessel lumen with impaired NO bioavailability [7]. Such endothelial dysfunction is the initiating step towards the development of atherosclerosis, a major cause of mortality and morbidity accounting for up to 31% of deaths worldwide in 2015 [8]. Impairment of the endothelial function is therefore recognized as the prequel to any cardiovascular and metabolic disease associated with hypertension, atherosclerosis, diabetes, hypercholesterolemia [9].
Since NO plays a major role in the maintenance of endothelial function, early detection of its reduced bioavailability would be of great interest to prevent cardiovascular events. Nevertheless, an easy quantification of NO in circulating blood is precluded by its very short half-life (approximately 2 milliseconds) related to its chemical reactivity, itself influenced by the redox status in the vasculature. A reliable assay of the bioavailability of NO in human circulation in vivo would be of high interest both for the screening of high-risk, yet asymptomatic cardiovascular patients and for the monitoring of existing endothelial dysfunction. Such an assessment would also be of interest as a surrogate biomarker to quantitatively measure the effects of new drugs developed to treat endothelial dysfunction.
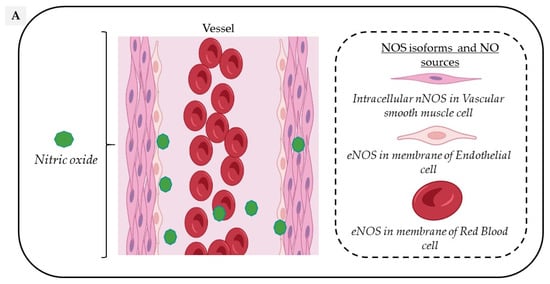
2. Vascular Sources of NO
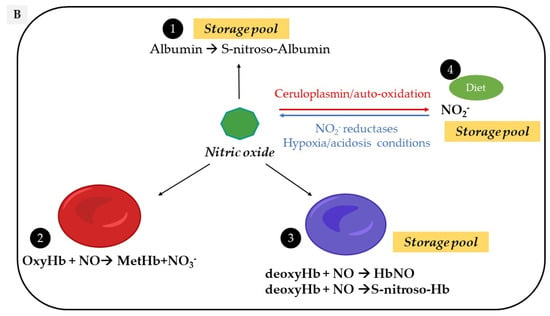
Circulating NO is mainly regulated through the activity of endothelial nitric oxide synthase (eNOS) but also from the nitrite-dependent NO synthesis, while nitroso/nitrosyl species in biological compounds can also act as a reservoir in the bloodstream (Figure 1).
Figure 1. Vascular sources and “reservoirs” of NO. (A) NO, produced by eNOS, diffuses to VSMC and the lumen of vessels. (B) In the lumen, NO reacts with albumin to form S-nitroso-albumin (1), with OxyHb to form MetHb and nitrate in normoxic conditions (2) and with deoxy-Hb to form HbNO (3) and S-nitroso-Hb in hypoxic conditions (3). S-nitroso-albumin (1) and S-nitroso-Hb (3) serve as storage pools of NO. NO can also be oxidized into nitrite under normoxic condition (4). Combined with the dietary intake, nitrite, in turn, it serves as the storage pool and can be reduced to NO under hypoxic and acidic conditions (4). In VSMC, NO is also produced by nNOS. deoxy-Hb: deoxygenated hemoglobin; EC: endothelial cells, eNOS: endothelial Nitric Oxide synthase; HbNO: nitrosylated hemoglobin; MetHb: methemoglobin; nNOS: neuronal Nitric Oxide synthase; NO: nitric oxide; NO2−: nitrite; NO3−: nitrate; OxyHb: oxygenated hemoglobin; RBC: red blood cells; S-nitroso-Hb: S-nitroso-hemoglobin; VSMC: vascular smooth muscular cells.
2.1. Oxygen-Dependent Nitric Oxide Synthesis
In the vascular system, NO is mainly produced from the conversion of L-Arginine to L-Citrulline by the constitutive vascular endothelial NOS isoform (eNOS). However, all three NOS isoforms are potentially expressed in various cell types composing the vascular tissue and act in a coordinated manner through their appropriate cellular–subcellular localization. In blood vessels, eNOS is mainly localized in the caveolae of endothelial cells, while the neuronal NOS (nNOS) is expressed in the sarcoplasmic reticulum from vascular smooth muscle cells (VSMC); in addition, the inducible NOS (iNOS) is expressed in the cytosol of all cell types upon exposure to inflammatory cytokines such as Interleukin 1 beta (IL1-beta) and Interferon-gamma. Both eNOS and nNOS are constitutively expressed but are strongly regulated by transcriptional, post-transcriptional and post-translational mechanisms, including S-glutathionylation, S-nitrosation (which both inhibit its function) or protein–protein interactions, such as the inhibitory interaction of eNOS with caveolin-1 at the plasma membrane [10].
Both these enzymes constantly produce small amounts of NO in the vessel wall and in the lumen—particularly eNOS, which is activated by shear stress and stretch [11]. In contrast, the inducible iNOS, once expressed, produces much higher amounts of NO that is mainly involved in nitrosative reactions associated with pathological vascular remodeling [12].
The functional eNOS is organized as a homodimer and needs oxygen and cofactors such as reduced nicotinamide-adenine-dinucleotide phosphate (NADPH), Flavin adenine dinucleotide (FAD), Flavin mononucleotide (FMN) and tetrahydro-L-biopterin (BH4) to be catalytically active [13]. Full eNOS activation requires the binding of Ca2+, Calmodulin and a number of kinases in order to phosphorylate the enzyme on Ser1177, its main activation site (e.g., by protein kinase A, B, C, G, AMP-activated protein kinase and Calmodulin-Dependent Protein Kinase II) and phosphatases to dephosphorylate the negative regulatory site on Thr495 [14]. Under physiological circumstances, and as long as eNOS remains dimeric, NO signaling participates to the vascular homeostasis through a cyclic guanosine-5-monophosphate (cGMP)-dependent or via a cGMP-independent pathway [15,16].
eNOS is mainly expressed in endothelial cells, but is also found in platelets [17] and erythrocytes [18]. Endothelial eNOS-derived NO appears as the central regulator of vascular homeostasis by playing crucial roles both in the vascular wall by relaxing and inhibiting VSMC hypertrophy, promoting angiogenesis, but also by diffusion into the vessel lumen where it inhibits leukocyte adhesion, platelet aggregation and thrombosis. An additional involvement in vascular relaxation is provided by nNOS through an Angiotensin II- dependent stimulation mainly in hypertension [19].
Through a functional, calcium-dependent, eNOS-like enzyme, erythrocytes also produce, store and distribute NO in peripheral tissues with a potential role in hypoxia-induced vasodilation [20], platelet inhibition [21] and regulation of red blood cells (RBC) deformability and aggregation [22]. While some contradictory results have questioned the specific role of erythrocyte NOS [23], its involvement is supported by decreased erythrocyte-released NO metabolites (nitrite and nitrate) levels in response to NOS inhibition [24]. A specific contribution of erythrocyte eNOS to blood pressure regulation and nitrite homeostasis has gained recognition from experiments using bone marrow transplantation in chimeric mice competent or deficient for eNOS expression in circulating blood cells [25]. Erythrocyte eNOS expression and function have also emerged as independent risk factors of cardiovascular disease [26].
2.2. Nitrite-Dependent Nitric Oxide Synthesis
An alternative pathway can contribute additional NO from the inorganic anions, nitrate (NO3−) and nitrite (NO2−). Diet, particularly from vegetables, is the main provider of exogenous nitrate [27] and from cured meats (∼30%), of nitrite, to a lesser extent [28]. Nitrate is reduced into nitrite through the nitrate reductase activity of the bacteria in the oral cavity and enters the stomach where it undergoes an acid-dependent reduction into nitrous acid (HNO2) [27]. Nitrous acid then decomposes into nitrogen species, which are partially absorbed into the circulation and recycled to form NO or other bioactive nitrogen species. The reduction of nitrite to NO is enhanced by ascorbic acid and polyphenols, both reducing compounds commonly found in vegetables and fruits. After dietary intake, under normoxic conditions, the L-Arginine-NOS pathway is the main supplier of NO oxidation products from the rapid oxidation of NO by heme proteins (e.g., hemoglobin, to form nitrate with production of methemoglobin) and by cytochrome c oxidase and ceruloplasmin (to form nitrite) [29]. In addition, NO auto-oxidation can also occur [30,31]. Nitrite is considered as a reservoir of NO that can be reduced to bioactive NO through the nitrite reductase activity of a number of proteins, particularly under hypoxic and acidic conditions [28], while hemoglobin and myoglobin would act as oxygen sensors. This mechanism may therefore serve as a backup system mainly when oxygen-dependent NOS function is limited [32].
2.3. Nitroso/Nitrosyl Species
As a free radical, the NO “nitrosyl” moiety can easily react either with carbon, nitrogen, oxygen or sulfur atoms, resulting in a nitrosation reaction (where the incorporated NO is referred to as a nitroso group) [33]. NO reaction with a metal-centered protein (e.g., the catalytic site of a metalloenzyme) will induce a nitrosylation reaction (where the incorporated NO is referred to as nitrosyl group) [34]. Protein S-nitrosation (R-SNO) with the thiol group of a cysteine residue has emerged as a main posttranslational modification involved in the regulation of cell signaling proteins (including eNOS as reported above) while repeated transnitrosation reactions enable the incorporation of NO groups into other organic molecules [35]. These compounds naturally occur in vivo; stabilize NO; potentiate its biological effects and mostly act as a storage pool for NO (particularly S-nitroso-Albumin, S-nitroso-Hemoglobin and S-nitrosated proteins) [36].
2.4. Red Blood Cells
Erythrocytes have been considered as “NO sinks” due to different interactions be-tween NO and hemoglobin depending on its state. Hemoglobin exists in two distinct conformational states with less affinity for oxygen in the T-state (tense) than the R-state (relaxed). The T-state corresponds to the deoxy-form of hemoglobin and is also known as deoxyhemoglobin (deoxy-Hb) [37]. A dioxygenation reaction in which NO reacts with oxygenated hemoglobin (oxy-Hb) induces the formation of inactive methemoglobin and nitrate, which cannot be converted back to nitrite in the circulation. An interaction of NO with deoxy-Hb produces both iron-nitrosyl hemoglobin (NO bound to the heme group; HbNO) and S-nitroso hemoglobin (NO bound to the thiol group of cysteine 93 in the β-globin chain) [38,39]. These two reactions have both high association and slow dissociation rates supporting the theory of a “NO sink” in erythrocytes and hemoglobin as scavenger of NO [38,40]. However, in vivo, pressures gradients push erythrocytes into the center of the blood vessel, creating a cell-free zone that attenuates the interaction between endothelial cells, NO and hemoglobin, thereby limiting scavenging [41]. Moreover, NO consumption by erythrocytes is 1000 times slower than free hemoglobin, suggesting intrinsic factors limiting NO consumption, including extracellular diffusion limitations [42]. In addition, two pathways have been suggested to mediate the export of NO from the erythrocytes via band 3, either in a metabolon complex with deoxyhemoglobin promoting its nitrite reductase activity [43] or by band 3-specific transnitrosation from S-nitroso-hemoglobin to other NO acceptors outside the cell [44].
These propositions combined with the production of NO from the erythrocyte NOS and of a significant amount of NO derivatives (including S-nitrosothiols and S-nitroso-hemoglobin) preserving NO from binding to oxyhemoglobin argue against the NO “scavenging” in erythrocytes.
Thus, erythrocytes should rather be considered as NO transporters reflecting vascular NO contents and, in a limited fashion, as NO generators through their eNOS activity, albeit with marginal influence on erythrocyte NO content.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27227921
This entry is offline, you can click here to edit this entry!