Uric acid is the end-product of purine metabolism in humans and apes, unlike other mammals which have uricase. This genetic evolution has led humans to exhibit plasma uric acid levels that are 3–10 times higher than those of other mammals. The association of hyperuricemia with increased cardiovascular risk may partly be explained by the activation of the renin-angiotensin system (RAS). When mild hyperuricemia was induced in rats by providing oxonic acid in the diet, blood pressure was elevated, and juxtaglomerular renin expression increased. Plasma renin activity and plasma aldosterone concentration were also elevated in rats with hyperuricemia, but these associations were unclear in adults with essential hypertension. Intrarenal RAS activity may be affected by hyperuricemia in humans.
- autosomal dominant tubulointerstitial kidney disease
- chronic kidney disease
- diabetes mellitus
- thiazide
- loop diuretic
1. Asymptomatic Hyperuricemia in Chronic Kidney Disease (CKD)
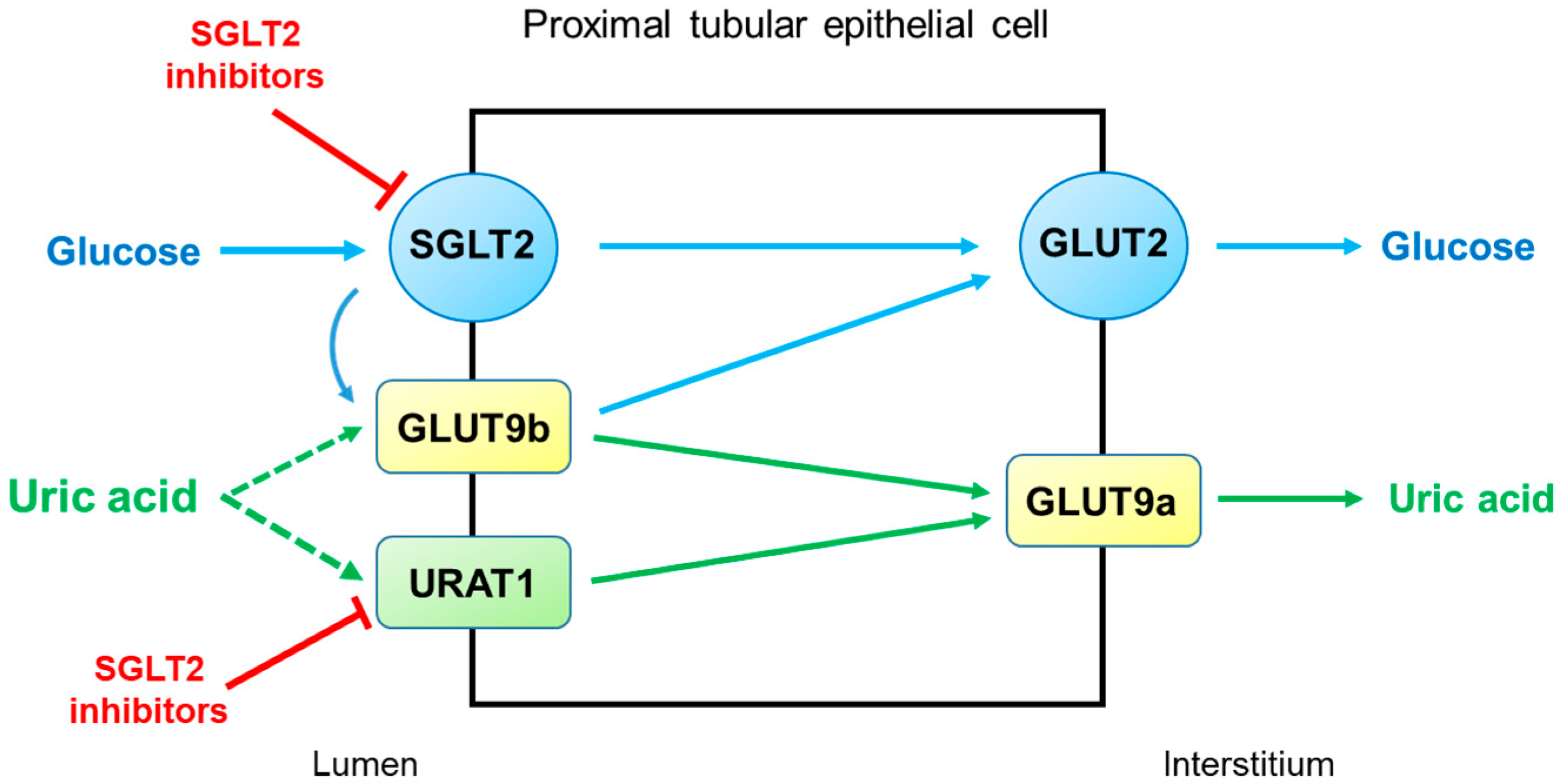
2. Diabetic Kidney Disease (DKD)
3. Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD)
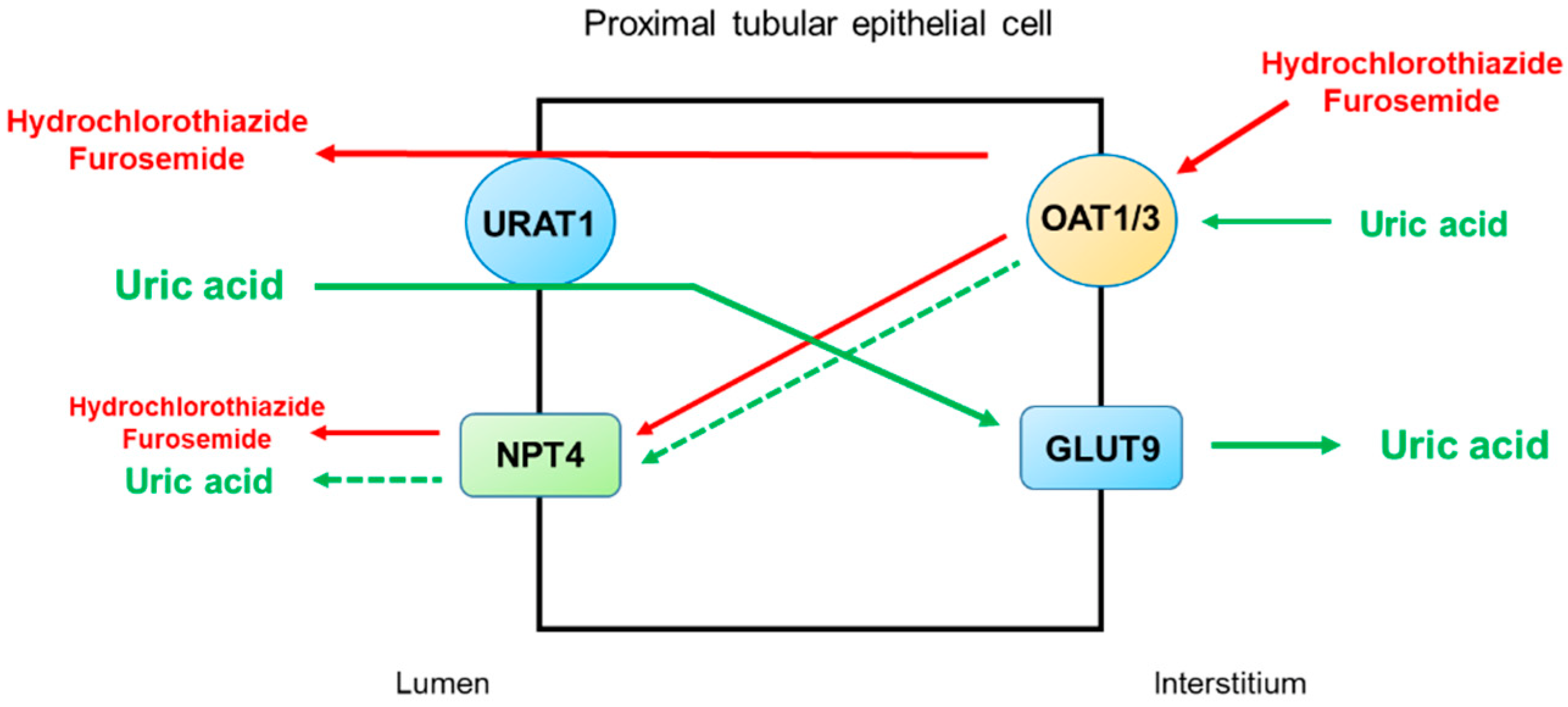
4. Thiazide and Loop Diuretics
This entry is adapted from the peer-reviewed paper 10.3390/life12111891
References
- Gonçalves, D.L.N.; Moreira, T.R.; da Silva, L.S. A systematic review and meta-analysis of the association between uric acid levels and chronic kidney disease. Sci. Rep. 2022, 12, 6251.
- Floege, J.; Johnson, R.J. Hyperuricemia and progression of chronic kidney disease: To treat or not to treat? Kidney Int. 2021, 99, 14–16.
- Badve, S.V.; Pascoe, E.M.; Tiku, A.; Boudville, N.; Brown, F.G.; Cass, A.; Clarke, P.; Dalbeth, N.; Day, R.O.; de Zoysa, J.R.; et al. Effects of allopurinol on the progression of chronic kidney disease. N. Engl. J. Med. 2020, 382, 2504–2513.
- Doria, A.; Galecki, A.T.; Spino, C.; Pop-Busui, R.; Cherney, D.Z.; Lingvay, I.; Parsa, A.; Rossing, P.; Sigal, R.J.; Afkarian, M.; et al. Serum urate lowering with allopurinol and kidney function in type 1 diabetes. N. Engl. J. Med. 2020, 382, 2493–2503.
- Jordan, D.M.; Choi, H.K.; Verbanck, M.; Topless, R.; Won, H.H.; Nadkarni, G.; Merriman, T.R.; Do, R. No causal effects of serum urate levels on the risk of chronic kidney disease: A Mendelian randomization study. PLoS Med. 2019, 16, e1002725.
- Sircar, D.; Chatterjee, S.; Waikhom, R.; Golay, V.; Raychaudhury, A.; Chatterjee, S.; Pandey, R. Efficacy of febuxostat for slowing the GFR decline in patients with CKD and asymptomatic hyperuricemia: A 6-month, double-blind, randomized, placebo-controlled trial. Am. J. Kidney Dis. 2015, 66, 945–950.
- Sezai, A.; Soma, M.; Nakata, K.; Osaka, S.; Ishii, Y.; Yaoita, H.; Hata, H.; Shiono, M. Comparison of febuxostat and allopurinol for hyperuricemia in cardiac surgery patients with chronic kidney disease (NU-FLASH trial for CKD). J. Cardiol. 2015, 66, 298–303.
- Johnson, R.J.; Nakagawa, T.; Jalal, D.; Sánchez-Lozada, L.G.; Kang, D.H.; Ritz, E. Uric acid and chronic kidney disease: Which is chasing which? Nephrol. Dial. Transpl. 2013, 28, 2221–2228.
- Hassan, W.; Shrestha, P.; Sumida, K.; Thomas, F.; Sweeney, P.L.; Potukuchi, P.K.; Rhee, C.M.; Streja, E.; Kalantar-Zadeh, K.; Kovesdy, C.P. Association of uric acid-lowering therapy with incident chronic kidney disease. JAMA Netw. Open. 2022, 5, e2215878.
- Otani, N.; Ouchi, M.; Misawa, K.; Hisatome, I.; Anzai, N. Hypouricemia and urate transporters. Biomedicines 2022, 10, 652.
- Facchini, F.; Chen, Y.D.; Hollenbeck, C.B.; Reaven, G.M. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991, 266, 3008–3011.
- Ji, P.; Zhu, J.; Feng, J.; Li, H.; Yu, Q.; Qin, H.; Wei, L.; Zhang, J. Serum uric acid levels and diabetic kidney disease in patients with type 2 diabetes mellitus: A dose-response meta-analysis. Prim. Care Diabetes 2022, 16, 457–465.
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2018, 20, 458–462.
- Bailey, C.J. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes. Metab. 2019, 21, 1291–1298.
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404.
- Novikov, A.; Fu, Y.; Huang, W.; Freeman, B.; Patel, R.; van Ginkel, C.; Koepsell, H.; Busslinger, M.; Onishi, A.; Nespoux, J.; et al. SGLT2 inhibition and renal urate excretion: Role of luminal glucose, GLUT9, and URAT1. Am. J. Physiol. Ren. Physiol. 2019, 316, F173–F185.
- Suijk, D.L.S.; van Baar, M.J.B.; van Bommel, E.J.M.; Iqbal, Z.; Krebber, M.M.; Vallon, V.; Touw, D.; Hoorn, E.J.; Nieuwdorp, M.; Kramer, M.M.H.; et al. SGLT2 Inhibition and uric acid excretion in patients with type 2 diabetes and normal kidney function. Clin. J. Am. Soc. Nephrol. 2022, 17, 663–671.
- Mabillard, H.; Sayer, J.A.; Olinger, E. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease. Nephrol. Dial. Transpl. 2021, 98, gfab268.
- Devuyst, O.; Olinger, E.; Weber, S.; Eckardt, K.U.; Kmoch, S.; Rampoldi, L.; Bleyer, A.J. Autosomal dominant tubulointerstitial kidney disease. Nat. Rev. Dis. Prim. 2019, 5, 60.
- Bleyer, A.J.; Kmoch, S. Autosomal dominant tubulointerstitial kidney disease: Of names and genes. Kidney Int. 2014, 86, 459–461.
- Bleyer, A.J.; Kidd, K.; Živná, M.; Kmoch, S. Autosomal dominant tubulointerstitial kidney disease. Adv. Chronic Kidney Dis. 2017, 24, 86–93.
- Scolari, F.; Caridi, G.; Rampoldi, L.; Tardanico, R.; Izzi, C.; Pirulli, D.; Amoroso, A.; Casari, G.; Ghiggeri, G.M. Uromodulin storage diseases: Clinical aspects and mechanisms. Am. J. Kidney Dis. 2004, 44, 987–999.
- Liu, Y.; Goldfarb, D.S.; El-Achkar, T.M.; Lieske, J.C.; Wu, X.R. Tamm-Horsfall protein/uromodulin deficiency elicits tubular compensatory responses leading to hypertension and hyperuricemia. Am. J. Physiol. Renal. Physiol. 2018, 314, F1062–F1076.
- Stavrou, C.; Koptides, M.; Tombazos, C.; Psara, E.; Patsias, C.; Zouvani, I.; Kyriacou, K.; Hildebrandt, F.; Christofides, T.; Pierides, A.; et al. Autosomal-dominant medullary cystic kidney disease type 1: Clinical and molecular findings in six large Cypriot families. Kidney Int. 2002, 62, 1385–1394.
- Okorn, C.; Goertz, A.; Vester, U.; Beck, B.B.; Bergmann, C.; Habbig, S.; König, J.; Konrad, M.; Müller, D.; Oh, J.; et al. HNF1B nephropathy has a slow-progressive phenotype in childhood-with the exception of very early onset cases: Results of the German Multicenter HNF1B Childhood Registry. Pediatr. Nephrol. 2019, 34, 1065–1075.
- Bingham, C.; Hattersley, A.T. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol. Dial. Transpl. 2004, 19, 2703–2708.
- Ferrè, S.; Veenstra, G.J.; Bouwmeester, R.; Hoenderop, J.G.; Bindels, R.J. HNF-1B specifically regulates the transcription of the γa-subunit of the Na+/K+-ATPase. Biochem. Biophys. Res. Commun. 2011, 404, 284–290.
- Ben Salem, C.; Slim, R.; Fathallah, N.; Hmouda, H. Drug-induced hyperuricaemia and gout. Rheumatology 2017, 56, 679–688.
- Palmer, B.F. Metabolic complications associated with use of diuretics. Semin. Nephrol. 2011, 31, 542–552.
- Pascual, E.; Perdiguero, M. Gout, diuretics and the kidney. Ann. Rheum. Dis. 2006, 65, 981–982.
- Jutabha, P.; Anzai, N.; Wempe, M.F.; Wakui, S.; Endou, H.; Sakurai, H. Apical voltage-driven urate efflux transporter NPT4 in renal proximal tubule. Nucleosides Nucleotides Nucleic Acids. 2011, 30, 1302–1311.
- Hamada, T.; Kuwabara, M.; Watanabe, A.; Mizuta, E.; Ohtahara, A.; Omodani, H.; Watanabe, M.; Nakamura, H.; Hirota, Y.; Miyazaki, S.; et al. A comparative study on the effectiveness of losartan/hydrochlorothiazide and telmisartan/hydrochlorothiazide in patients with hypertension. Clin. Exp. Hypertens. 2014, 36, 251–257.