Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
TRAIL is a type II transmembrane protein that undergoes proteolytic cleavage to produce an extracellular ligand. TRAIL can bind to decoy receptor 1 (DcR1) which lacks a DD altogether, and DcR2 which has a truncated DD. These decoy receptors are unable to induce DISC formation and act as negative regulators of the apoptotic signaling by competitively binding TRAIL. The canonical TRAIL-induced apoptotic signaling pathway is an example of apoptosis mediated through the extrinsic death pathway, which entails activation of cell-surface receptors by a ligand to induce activation of downstream caspases.
- TRAIL
- death receptors
- apoptosis
- triple-negative breast cancer
1. Introduction
Triple-negative breast cancer (TNBC) is defined by the absence of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2) amplification and accounts for ~15% of breast cancer cases [1]. It has the worst prognosis of the major subtypes of breast cancer due in part to the absence of well-defined molecular targets [1]. TNBC responds to chemotherapy, but metastatic relapse of the treated early stage is frequent, and there are no curative therapies in the advanced stage [1]. While new targeted therapies (e.g., trophoblast antigen 2 [trop2]-targeted antibody-drug conjugates [ADCs]) and immune checkpoint inhibitors have significant activity for the treatment of TNBC [2,3,4,5,6], novel therapies are still needed for TNBC especially in the advanced setting.
TRAIL is a member of the tumor necrosis factor (TNF) family of ligands capable of initiating apoptosis through engagement of its death receptors. TRAIL binds to Death Receptor 4 (DR4, also known as TRAIL-R1/TNFRSF10A) and Death Receptor 5 (DR5, also known as, TRAIL-R2/TRICK-2/KILLER/TNFRSF10B), causing caspase-mediated apoptosis (Figure 1A) [7,8]. TRAIL is mainly expressed on the cell surface of immune cells eliciting programmed death of target cells and has been reported to induce apoptosis in a variety of cancer cell lines while sparing the normal cells [7,9,10,11,12]. However, there are some reports of TRAIL-induced apoptosis in normal cells, such as primary salivary epithelial cells [13], prostate epithelial cells [14,15] and primary human epithelial esophageal cells [16]. Preclinical animal studies supported the lack of overt toxicity of TRAIL to the normal tissues [7,17,18]. Therefore, targeting TRAIL/DR pathway offers an attractive approach to induce apoptosis in cancer cells. Early attempts to treat cancer using the TNF and Fas ligand (FasL) as DR agonists showed disappointing results due to lack of efficacy or prohibitive preclinical toxicity [7,17,18]. This has prompted investigation into the use of TRAIL or DR4/5 agonist antibodies in cancer therapy. TRAIL agonist induced regression of cancer xenografts in mice without affecting normal tissues, and human phase 1 studies have demonstrated that TRAIL agonists are safe and well tolerated in patients [7,17,18,19,20,21,22,23]. However, phase 2/3 clinical trials utilizing recombinant human (rh)TRAIL and agonistic antibodies directed at DR4/5, while well-tolerated, have not shown significant clinical efficacy [23,24,25].
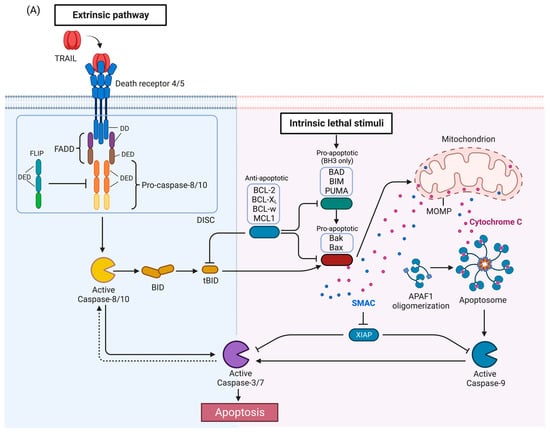
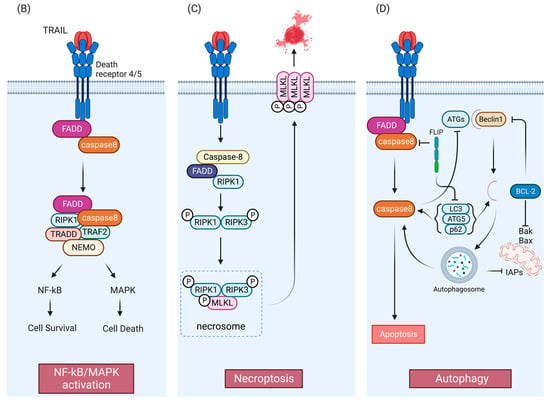
Figure 1. TRAIL signaling pathway. (A) Apoptosis pathway. Activation of DR4 and DR5 by TRAIL induces the extrinsic apoptosis pathway (left). The intrinsic pathway (right) is activated by a variety of stimuli and leads to release of proapoptotic proteins from the mitochondria. The two pathways interact as caspase-8 activated by the DRs can cleave BID which then activates the intrinsic pathway, and conversely, caspase-3 can cleave and activate caspase-8 in a feedback loop, thus amplifying the apoptotic signal. (B) TRAIL-mediated NF-kB/MAPK pathways. Anti- or pro-survival mechanisms appear to be context dependent. (C) TRAIL-mediated necroptosis pathway. (D) Crosstalk between TRAIL pathway and autophagy. APAF1, Apoptotic protease activating factor 1; DD, death domain; DED, death effector domain; DISC, death-inducing signaling complex; FADD, Fas-associated death domain protein; FLIP (FLICE [FADD-like IL-1β-converting enzyme]-inhibitory protein); MOMP, mitochondrial outer membrane permeabilization; SMAC, second mitochondrial activator of caspases; XIAP, X-linked inhibitor of apoptosis. TRAF2, TNF receptor-associated factor 2; NEMO, NF-kappa-B essential modulator; TRADD, Tumor necrosis factor receptor type 1-associated DEATH domain protein; NF-κB, nuclear factor-κB; MAPK, mitogen-activated protein kinase; MLKL, mixed-lineage kinase domain-like protein; ATG, Autophagy-related protein; LC3, Microtubule-associated proteins 1A/1B light chain 3; IAP: inhibitor of apoptosis. Figures were created with BioRender.com (accessed on 8 November 2022).
2. TRAIL Signaling Pathways
Activation of TRAIL death receptors by their cognate ligands induces apoptosis but as will be described below, under certain circumstances, activation of the TRAIL death receptors can promote growth and in tumors induce metastasis or pro-tumorigenic immune effects. The canonical TRAIL-induced apoptotic signaling pathway is an example of apoptosis mediated through the extrinsic death pathway, which entails activation of cell-surface receptors by a ligand to induce activation of downstream caspases (Figure 1A) [30]. Activation of DR4 and DR5 by TRAIL promotes receptor clustering and formation of the DISC [46]. The adaptor protein, Fas-associated death domain protein (FADD), also contains a death domain (DD) which interacts with the DD of DR4/5 in a homotypic fashion [46]. FADD also contains a death effector domain (DED) which recruits pro-caspases-8 and -10 via a homotypic interaction with DED of the pro-caspases [46]. The forced proximity of the pro-caspases-8 and -10 at the DISC leads to auto-processing of the pro-caspases [46], resulting in an active tetramer of two large and small subunits [46]. This results in activation of downstream caspases such as caspase-3 or -7, ultimately inducing apoptosis. FLIP (FLICE [FADD-like IL-1β-converting enzyme]-inhibitory protein), a negative regulator of the TRAIL pathway, is structurally related to pro-caspases-8 and -10 with N-terminal DED domains, and a C-terminal caspase-like domain, but the catalytic cysteine is replaced by tyrosine, thus rendering it catalytically inactive [47]. FLIP may also be recruited to the DISC, prevent caspase-8 or caspase-10 from interacting with FADD, and thus attenuate the apoptotic signal [46,47].
In some cells, activated caspase-8 or -10 can cleave Bid, a pro-apoptotic BH3-only BCL-2 family protein, into truncated Bid (tBid), which translocate to the mitochondria and activates the intrinsic death pathway (Figure 1A) [48,49,50]. The BCL-2 protein family regulates the intrinsic death pathway via anti- and pro-apoptotic family members [49]. When the intrinsic death pathway is activated, pro-apoptotic BCL-2 family members, such as BAD, BIM, and PUMA, antagonize anti-apoptotic family members, including BCL-2, BCL-xL, BCL-w, and MCL1 [49,51]. tBid directly and indirectly activates pro-apoptotic proteins Bak and Bax, causing mitochondrial outer membrane permeabilization (MOMP) and cytochrome c release [34,48,49,52]. The scaffold protein apoptotic protease-activating factor 1 (APAF1) binds to cytochrome c and activates caspase-9 [48,49]. Second mitochondrial activator of caspases (SMAC), an inhibitor of apoptosis proteins (IAPs) that suppress caspase function, is also released during MOMP to help facilitate apoptosis [53]. The extrinsic and intrinsic death pathways converge; caspases-8, -10, and -9 mediate proteolytic processing of the executioner caspases-3 and -7 which carry out the final steps of apoptosis by cleaving numerous substrates [48,49]. Activated caspase-3 is also able to activate caspase-8 in a feedback-loop, thus amplifying the apoptotic signal [54,55].
Like TNF, TRAIL-induced DR4/5 activation has also been associated with the induction of nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling [47,56,57,58] (Figure 1B). TNF signaling is a more potent activator of these pathways than TRAIL [57]. After TRAIL-induced caspase activation, receptor-interacting serine/threonine-protein kinase 1 (RIPK1/RIP1), FADD, TNF receptor-associated factor 2 (TRAF2), and caspase-8 form a secondary complex that facilitates kinase signaling in a RIPK1-dependent manner [57]. However, findings have been mixed concerning the anti- and pro-survival mechanisms induced by TRAIL-mediated NF-κB and MAPK activation [40,59]. Inhibition of components of the NF-κB and MAPK signaling pathways has produced examples of both enhanced and attenuated sensitivity to TRAIL-induced apoptosis, demonstrating that the outcomes associated with TRAIL-mediated activation of NF-κB and MAPK will vary depending on context and requires further characterization [40,59].
TRAIL signaling is also associated with non-apoptotic cell death mechanisms, including the caspase-independent, cell-regulated form of necrosis, necroptosis [59,60,61,62,63] (Figure 1C). TRAIL has been found to induce necroptosis regulators RIPK1 and RIPK3 in an acidic pH-dependent manner, and NF-κB inhibition has been found to enhance sensitivity to TRAIL-induced necroptosis [59,60,62,63]. Mechanistically, necroptosis depends on activation of RIPK1 and RIPK3 and necrosome complex formation involving RIPK1 and RIPK3 and the mixed lineage kinase domain-like protein (MLKL) [64] (Figure 1C). Recently, a study indicated the involvement of an E3 ubiquitin-protein ligase TRIM21(tripartite motif containing 21) in endogenous TRAIL-mediated necrosome formation [65]. Further elucidation of the mechanisms and conditions under which TRAIL activates necroptotic signaling is needed.
Importantly, numerous studies show that TRAIL can also induce autophagy [66]. Autophagy and apoptosis are both important cellular processes controlled by distinct groups of regulatory mechanisms [67]. They also have a crosstalk to regulate each other [68,69,70]. As shown in Figure 1D, autophagy and apoptosis share same regulatory factors, including the Bcl-2 family [49,69,71] and FLIP [72]. Bcl-2 family members inhibit Beclin-1 [73,74] which is required for activation of autophagy [75]. FLIP limits the ATG3-mediated LC3 conjugation to inhibit autophagosome biogenesis [72]. Caspase-8 has been shown to regulate autophagy by targeting autophagic components, such as ATG3, ATG5 and Beclin-1 [76,77,78,79]. Autophagosome formation can facilitate caspase-8 activation by providing a platform consisting of ATG5 and LC3 in some contexts [80], while sequestration of pro-caspase-8 to the autophagosome can lead to either caspase-8 induction, or downregulation [81]. The importance of autophagy in TRAIL resistance breast cancer cells is still being studied intensively. Notably, in different models of breast cancer cell lines resistance to TRAIL-induced killing was attributed by TRAIL induced autophagy [82]. Furthermore, in a series of breast cancer cell lines autophagy was shown to have an impact on dynamics of TRAIL receptors. In these cell lines, autophagosome accumulation led to decrease in TRAIL-induced death by downregulating the TRAIL receptors [83]. While these data suggest that TRAIL-induced autophagy inhibits TRAIL-mediated death, there are descriptions of TRAIL-induced autophagic cell death. For example, in a 3D culture model of breast lumen formation using the immortalized but not transformed human mammary MCF-10A cell line, TRAIL-induced autophagy contributed to the death of the luminal cells that led to hollow lumen formation [84]. Thus, understanding TRAIL-induced autophagy is required to decipher TRAIL mediated response or resistance.
TRAIL and DR expression have been shown to positively regulate cell growth under certain conditions. Specifically, TRAIL signaling promotes proliferation and IFNγ production in pre-activated T cells [85]. TRAIL-induced activation of DR5 and mTRAIL-R signaling is correlated with enhanced TNBC-associated bone metastasis and KRAS-driven lung and pancreatic cancer metastasis, respectively [86,87]. The first study utilized a bone tropic variant of the TNBC MDA-MB-231 cell line which is known to contain an oncogenic KRAS mutation which is infrequent in breast cancer [88]. In this study the investigators found that DR5 was upregulated in the bone tropic cells compared to the parental MDA-MB-231 cell line and they demonstrate that RNAi mediated knockdown of DR5 reduces the levels of bone metastasis related genes (e.g., HMGA2, phosphor-Src, and the C-X-C cytokine receptor 4) in the cancer cell. When injected intracardially, the knockdown of DR5 reduced the ability of the cells to metastasize to bone [86]. Thus, the pro-metastatic phenotypes were reversed with DR5 and mTRAIL-R inhibition, suggesting that inhibition of the TRAIL signaling may provide a novel method to inhibit metastasis.
As discussed above, TRAIL knockout mice have shown increased metastases [45]. Additional characterization of the conditions under which TRAIL signaling is inhibitory or pro-proliferative or pro-metastatic is necessary to determine the contexts wherein TRAIL pathway activation or inhibition are the appropriate therapeutic strategy in TNBC.
This entry is adapted from the peer-reviewed paper 10.3390/cells11233717
This entry is offline, you can click here to edit this entry!