The Michael addition was named after Arthur Michael (1853–1942) [
1] who discovered the addition reaction of sodiomalonate esters or sodioacetoacetate esters onto α,β-unsaturated esters [
2]. This reaction corresponds to a 1,4-conjugate addition of a stabilized carbanion onto an electron-poor double bond such as α,β-unsaturated carbonyl compounds. Other Michael additions based on the addition of an anion to activated alkenes have been further developed by using other nucleophiles. Thia-, oxa- and aza-Michael reactions correspond, respectively, to the polar 1,4-addition of a thiol, an alcohol and an amine (Michael donor) onto double bonds activated by a conjugated electron-withdrawing group (Michael acceptor) [
3]. In contrast to the historical Michael reaction, these “hetero-Michael” additions are potentially reversible under appropriate conditions.
Covalent adaptable networks (CANs) have gained a lot of interest from the scientific community, as this polymer family represents a potentially sustainable solution for the reduction of polymer wastes. Indeed, thermosets, which were originally designed to be dimensionally stable and provide high mechanical and chemical resistance, cannot be easily reprocessed [
4]. CANs are composed of a 3D network structure in which some covalent bonds show reversibility upon thermal stimulus, conferring self-healing, shape memory and reprocessing properties to the material [
5]. The first CANs, composed of disulfide bonds and silyl ethers, have been developed decades ago [
6,
7] and recently re-evaluated [
8,
9]. Numerous exchange or reversible reactions such as Diels–Alder cycloaddition [
10,
11,
12,
13], transesterification [
14,
15,
16,
17], transcarbamoylation [
18,
19,
20,
21] or transamidation [
22,
23] have been employed to design and prepare CANs.
The exchange reactions are generally classified according to their mechanism, either associative or dissociative [
24]. Based on this mechanistic difference, vitrimers would correspond to CANs having an associative exchange mechanism, whereas the exchange reaction will follow a dissociative mechanism in dissociative CANs. An associative exchange ensures a constant cross-link density, whereas a dissociative one generates a temporary loss of network connectivity when the exchange takes place. Therefore, only associative networks should show an Arrhenius behavior and therefore be considered as vitrimers (associative CANs). Nevertheless, as highlighted by Dichtel and Elling [
25], some dissociative CANs based on oxime-enabled transcarbamoylation [
26] and
trans-N-alkylation [
27] for instance also showed this typical behavior. Hence, the major difference between associative and dissociative CANs is often a decrease of the storage modulus with temperature as observed in different studies on dissociative CANs [
22,
28].
2. Thia-Michael Reaction
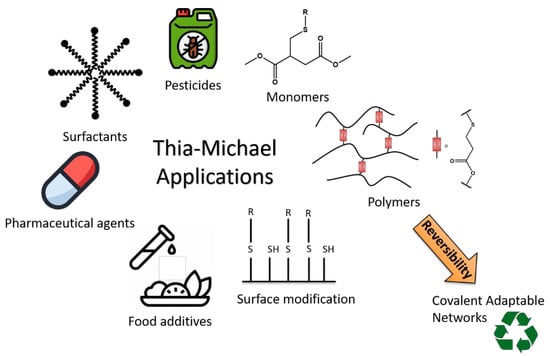
The thia-Michael addition is an intensively used reaction in industry for the synthesis of food additives and surfactants, pesticides and pharmaceutical agents [
34,
35,
36,
37,
38]. In recent decades, the thia-Michael addition has found a new field of application in materials science. Indeed, the simplicity of this addition reaction enables to easily synthesize a large range of monomers [
39] or dendrimers [
40] and can also be used to perform surface modification [
41]. Polymer networks synthesized via thia-Michael addition have been also explored. For instance, thio-acrylate networks were prepared by using a thia-Michael addition followed by the photopolymerization of an excess of acrylate functional groups [
42]. Using a similar idea, A. Lowe et al. performed sequential phosphine-catalyzed thia-Michael addition followed by radical-mediated thiol−yne reaction to prepare polyfunctional materials [
43]. The thia-Michael addition has also been used to cross-link acrylated epoxidized soybean oil in order to obtain partially bio-based networks [
44]. Recently, some dynamic covalent networks (CANs) have been designed on the basis of the reversibility of the thia-Michael addition (
Scheme 1) [
45,
46]. For the following discussion, the term “thia-Michael addition” refers only to the addition step, whereas the term “thia-Michael equilibrium” refers to the overall thia-Michael reaction (elimination/addition). Finally, the term “thia-Michael exchange” refers to the reaction between a Michael adduct and a Michael donor or acceptor. The thia-Michael exchange, which is focused on in this review, is of course dependent on a few critical parameters such as the promotor or catalyst, the solvent and the structure of the reagents (thiol and Michael acceptor).
Scheme 1. Application fields of thia-Michael addition.
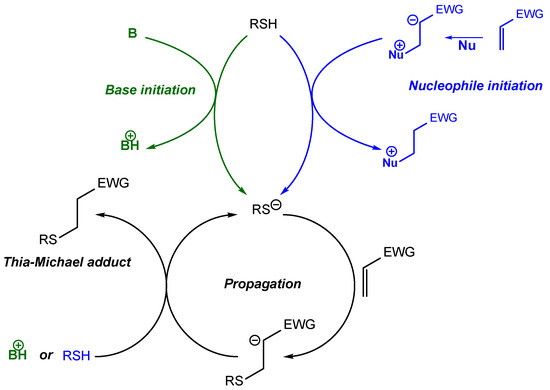
Influence of the promotor/catalyst: Thia-Michael additions are generally catalyzed with weak Brønsted bases such as triethylamine or by Lewis bases such as phosphines (nucleophile-initiation) [
47,
48]. Bowman et al., studied these two mechanisms [
49] and established that, in both cases, the addition takes place in three steps. First, a thiolate is formed by reaction of the thiol either with the base or with an anion generated from the nucleophilic catalyst and the Michael acceptor. Then, this thiolate adds to the acceptor to generate a negatively charged addition product. Finally, a proton exchange occurs between this addition product and either a new thiol function or the protonated base to form the final Michael adduct (
Scheme 2) [
49]. For the nucleophile-initiated thia-Michael addition, a zwitterionic intermediate is generally formed from the attack of the Lewis base onto the Michael acceptor. Subsequently, the thiol is deprotonated by the zwitterionic intermediate leading to the formation of the thiolate anion and a phosphonium ester. After addition of the thiolate onto the Michael acceptor, a proton exchange occurs with a new thiol, leading to a new thiolate and therefore to a propagation of the reaction. In this case, the nucleophile is only used as an initiator of the reaction. For the base-catalyzed mechanism, the thiolate anion is obtained in one step by acid-base reaction. In this case, the base and the unreacted thiol can both perform proton exchange with the negatively charged addition product, whereas in the nucleophile-initiated thia-Michael addition only the thiol species participate to this exchange. The base-catalyzed rate is negatively impacted by the presence of the protonated base in the system as it slows down the formation of new thiolate. Therefore, the nucleophile-initiated thia-Michael addition generally proceeds faster and requires lower catalyst loadings compared to the base-catalyzed pathway [
50].
Scheme 2. The mechanism of the base-catalyzed and nucleophile-initiated thia-Michael additions between a thiol and a vinyl group.
Influence of the solvent: The solvent used for a chemical reaction is generally chosen to solubilize the reactants and catalysts and to minimize side reactions [
3]. However, solvent characteristics also play a role in the reaction kinetics. For instance, the thiol-acrylate reaction catalyzed by triethylamine was accelerated in a polar medium such as dimethyl sulfoxide (DMSO), propylene carbonate and N-ethyl urethane compared to the neat reaction [
51]. Du Prez et al. investigated the Michael addition of thiol with maleimide, acrylate and vinyl sulfone and the NEt
3-catalyzed thiol–iso(thio)cyanate reactions in DMF and in chloroform [
52]. Overall, they observed higher reaction rates in polar aprotic solvents such as DMF and DMSO, which favor the formation of thiolates through a stabilization of this negatively charged species due to their high dielectric constants [
53]. Therefore, solvents promoting the formation of thiolates (initiation) should be chosen to promote the thia-Michael addition. This effect was particularly visible in the case of base-catalyzed reactions for which initial deprotonation is the limiting step of the mechanism. Similarly, addition reaction rates are increased in high pH solution as thiolate anions are easily formed under these conditions [
54].
Influence of the substrate structures: The thiol basicity and the electron deficiency of the Michael acceptor as well as the steric hindrance of the reactants influence the thia-Michael addition. The influence of the bulkiness of the substituents has been largely studied for the aza- and carbon-Michael additions, leading to the intuitive conclusion that the reaction rate decreases as the α- and β-substituents size increases [
55,
56]. Recently, Bowman et al., highlighted a similar behavior for thia-Michael additions with alkyl thiol. Indeed, when the steric hindrance of the thiol increases, the addition rate decreases. However, when mercaptopropionates were used, the addition of a methyl substituent in α-position of the thiol increased slightly the reaction rate, leading to the opposite conclusion [
57]. Nonetheless, as previously reported, the rate-limiting step is different between the addition of alkyl thiols or mercaptopropionates [
58]. Due to their respective basicity and nucleophilic characters, the addition of alkyl thiols is rate-limited by the proton exchange step whereas the mercaptopropionate reactions are limited by the thiolate addition step. Thus, the decrease of the deprotonation rate induced by the higher electron-density of substituted alkyl thiols, added to their increased steric hindrance, results in a reduction of the overall reaction rate [
59]. In contrast, the addition rate of mercaptopropionates is directly dependent on the thiol nucleophilicity and the most-electron-rich thiolate (α-methylmercatopropionate) adds faster onto the double bond. In this study, it was also demonstrated through a comparison between vinyl sulfone, fumarate and acrylate that the more electron-deficient the acceptor, the higher the addition rate. Tirelli et al. also showed that thia-Michael addition rate was higher for acrylates than for acrylamides, thus highlighting the electronegativity dependence of this reaction [
60].
The thia-Michael addition is highly efficient, rapid and more selective than the radical-mediated thiol-ene reactions [
61]. Indeed, the thiol-ene reaction involves the formation of a thiyl radical that directly adds onto the carbon-carbon double bond, yielding a carbon-centered radical intermediate. In a second step, an hydrogen transfer occurs with a second thiol molecule to form the thiol-ene addition product and a new thiyl radical [
53]. Secondary products are mainly formed through the homopolymerization of alkene radicals generated during the reaction [
62,
63]. In contrast to the radical-initiated thiol-ene reaction, the thia-Michael addition can be performed under neat conditions and at low temperature, with a higher tolerance to functionality [
50].
In summary, the thia-Michael addition can be considered as a performant “click” reaction that can be used in organic synthesis, surface modification and polymer chemistry [
64].
3. Thia-Michael Equilibrium and Exchange Kinetics
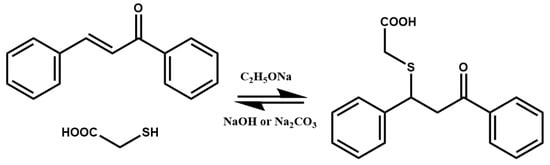
The reversible character of the thia-Michael addition was evidenced for the first time in 1931 by B. Nicolet [
65] who demonstrated the reversibility of the addition of thioglycolic acid on benzalacetophenone (
Scheme 3). The thia-Michael adducts dissociated when placed in a basic medium composed of sodium hydroxide or sodium carbonate.
Scheme 3. Thia-Michael equilibrium of thioglycolic acid on benzalacetophenone.
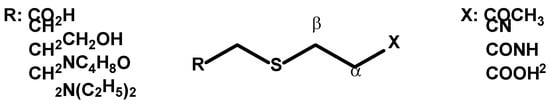
The dissociation phenomenon was then specifically evaluated by C. Allen and W. Humphlett [
66,
67]. The dissociation of thia-Michael adducts was monitored 2 min after dissolution in water or in ethanol with and without sodium hydroxide at different temperatures. The influence of the groups present on the acceptor or on the thiol was also examined in these articles (
Scheme 4). They demonstrated that the elimination reaction was favored as the electron withdrawing ability of the X group increased by enhancing the acidity of the α-hydrogen of the alkene function, as suggested by the order of dissociation observed in this study (ketone > nitrile > amide > acid). In contrast, the correlation between the structure of the thiol and the thia-Michael reversibility was however not obvious from these studies.
Scheme 4. Thia-Michael adducts.
In 1968, Holmes et al. [
68] observed the fast reaction of dually activated Michael acceptors with butanethiol. In this study, a cyano group is associated with another electron-withdrawing group on the α-position of the acceptor, leading to a fast addition reaction. However, the thia-Michael adducts could not be isolated due to the high reversibility of the reaction [
69,
70]. The dynamic character of the thia-Michael equilibrium is therefore largely influenced by the EWG placed in the α-position. This perspective has notably found applications in medicinal chemistry. The thia-Michael equilibrium indeed served as an application of the concept of reversible covalent inhibition, enabling the development of protein inhibitors with long-lasting, but not permanent action [
71]. This principle was specifically introduced by Taunton et al. who designed reversible Michael acceptors as cysteine-targeting inhibitors [
70,
72]. They highlighted the influence of a second EWG on α-cyano Michael acceptor with kinetic and computational analyses of the thia-Michael equilibrium. The presence of a methylthiazole group placed in the α-position of the acrylonitrile had a strong promoting impact on the reversibility of the thia-Michael addition, similar to the one observed with an α-amide group. This study also demonstrated that the presence of a phenyl group at the β-position did not have a significant effect on the reaction reversibility.
In 2016, Houk et al. also investigated the influence of an additional α-EWG and of a β-substituent by DFT calculations [
73]. They demonstrated that an α-EWG lowers the energy barrier for the thiol addition but also makes the addition reaction thermodynamically less favorable. The presence of an aryl or of a branched alkyl group on the β-carbon led to a rapidly reversible thia-Michael addition. The equilibrium of the thiol addition with benzalcyanoacetate-based Michael acceptors was specifically assessed by Herbert et al. [
74]. The equilibrium constants were determined at room temperature for a series of benzalcyanoacetate and were highly dependent of the electron-withdrawing/-donating ability of the substituent placed on the
para-position of the β-phenyl ring. Hence, as the electron-withdrawing ability increased, the equilibrium shifted preferentially to the associated state. By performing NMR in temperature, the authors demonstrated also that the equilibrium shifted to the dissociative state as the temperature increased.
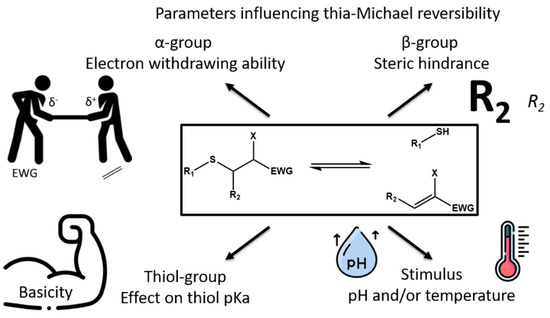
In conclusion of these first studies, three major parameters must be considered to control thia-Michael equilibrium: the α-EWG group, the stimulus used to activate the retro thia-Michael, and to a lesser extent the β-carbon substituents (
Scheme 6). Indeed, even if the thermal stimulus is the most common one, increase of the pH value or the presence of a base in the reaction medium also favor the retro-thia-Michael addition. For instance, thermodynamic and kinetic analyses of the thia-Michael equilibrium of glutathione onto helenalin derivatives were performed at different pH values [
75,
76]. It was demonstrated that the addition reaction proceeded rapidly and was reversible at basic pH, and that oppositely the reaction was slow and irreversible at low pH. The Michael addition of thiols onto 5-methylene pyrrolones has also been described as reversible in an alkali buffer (pH 9.5) and exchange reaction with other thiols was observed at 7.5 pH [
77].
Scheme 6. Parameters influencing thia-Michael equilibrium applications.