Aneuploidy is a hallmark of cancer and a major cause of miscarriages in humans. It is caused by chromosome segregation errors during cell divisions. Evidence is mounting that the probability of specific chromosomes undergoing a segregation error is non-random. In other words, some chromosomes have a higher chance of contributing to aneuploid karyotypes than others. This could have important implications for the origins of recurrent aneuploidy patterns in cancer and developing embryos. Here, we review recent progress in understanding the prevalence and causes of non-random chromosome segregation errors in mammalian mitosis and meiosis. We evaluate its potential impact on cancer and human reproduction and discuss possible research avenues.
- mitosis
- meiosis
- chromosomal instability
- non-random segregation errors
- cancer
- embryo
- development
- aneuploidy
1. Introduction
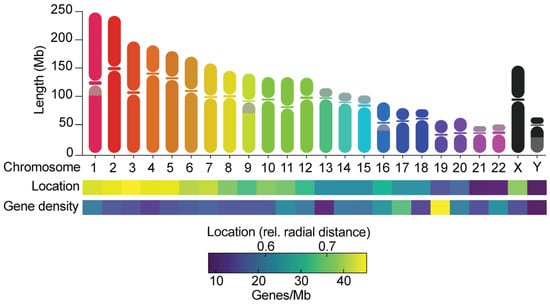
2. Evidence for Non-Random Chromosome Segregation Error Frequencies
2.1. In Mitosis
2.2. In Meiosis
3. Chromosome Segregation Mechanisms
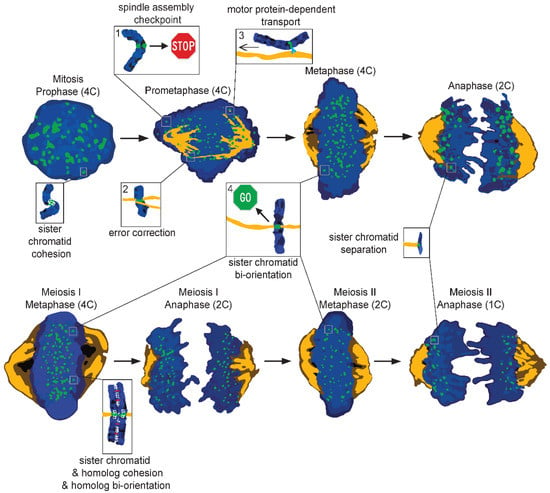
4. Mechanisms of Non-Random Chromosome Segregation Errors
4.1. Centromeric and Centromere-Proximal Features
4.2. Chromosome Size
4.3. Chromosome Location
5. Consequences of Non-Random Chromosome Segregation Errors
5.1. Karyotype Evolution in Cancer
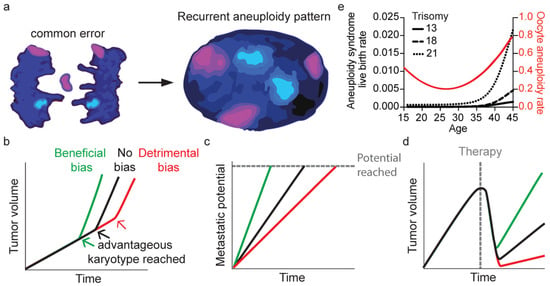
5.2. Aneuploid Live Births
6. Concluding Remarks and Future Directions
This entry is adapted from the peer-reviewed paper 10.3390/cells11223564
References
- Thompson, S.L.; Compton, D.A. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol 2008, 180, 665–672.
- Janssen, A.; Burg, M. Van Der; Szuhai, K.; Kops, G.J.P.L.; Medema, R.H. Chromosome Segregation Errors as a Cause of DNA Damage and Structural 2011, 333, 1895–1899.
- Bolhaqueiro, A.C.F.; Ponsioen, B.; Bakker, B.; Klaasen, S.J.; Kucukkose, E.; van Jaarsveld, R.H.; et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat Genet 2019, 51, 824–834.
- Knouse, K.A.; Wu, J.; Whittaker, C.A.; Amon, A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci 2014, 111, 13409–13414.
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; et al. Mosaic Copy Number Variation in Human Neurons. Science 2013, 342, 632–637.
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496.
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627.
- Starostik, M.R.; Sosina, O.A.; McCoy, R.C. Single-cell analysis of human embryos reveals diverse patterns of aneuploidy and mosaicism. Genome Res 2020, 30, 814–826.
- Hassold, T.J.; Jacobs, P.A. Trisomy in man. Annu Rev Genet 1984, 18, 69–97.
- McCoy, R.C.; Demko, Z.; Ryan, A.; Banjevic, M.; Hill, M.; Sigurjonsson, S.; et al. Evidence of Selection against Complex Mitotic-Origin Aneuploidy during Preimplantation Development. PLoS Genet 2015, 11, 1–31.
- Hoevenaar, W.H.M.; Janssen, A.; Quirindongo, A.I.; Ma, H.; Klaasen, S.J.; Teixeira, A.; et al. Degree and site of chromosomal instability define its oncogenic potential. Nat Commun 2020, 11, 1–11.
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.; Murphy, C.J.; Ly, P.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nat Publ Gr 2018, 553, 467–472.
- Diaz-Rodríguez, E.; Sotillo, R.; Schvartzman, J.; Benezra, R. Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo. Proc Natl Acad Sci 2008, 105, 16719–16724.
- Gordon, D.J.; Resio, B.; Pellman, D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012, 13, 189–203.
- Chunduri, N.K.; Menges, P.; Zhang, X.; Wieland, A.; Gotsmann, V.L.; Mardin, B.R.; et al. Systems approaches identify the consequences of monosomy in somatic human cells. Nat Commun 2021, 12, 1–17.
- Stingele, S.; Stoehr, G.; Peplowska, K.; Cox, J.; Mann, M.; Storchova, Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol 2012, 8, 1–12.
- Williams, B.R.; Prabhu, V.R.; Hunter, K.E.; Glazier, C.M.; Whittaker, C. a; Housman, D.E.; et al. Aneuploidy Affects Proliferation and Spontaneous Immortalization in Mammalian Cells. Science 2008, 322, 703–710.
- Passerini, V.; Ozeri-Galai, E.; de Pagter, M.S.; Donnelly, N.; Schmalbrock, S.; Kloosterman, W.P.; et al. The presence of extra chromosomes leads to genomic instability. Nat Commun 2016, 7, 1–12.
- Oromendia, A.B.; Dodgson, S.E.; Amon, A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev 2012, 26, 2696–2708.
- Sheltzer, J.M.; Blank, H.M.; Pfau, S.J.; Tange, Y.; George, B.M.; Humpton, T.J.; et al. Aneuploidy drives genomic instability in yeast. Science 2011, 333, 1026–1030.
- Torres, E.M.; Sokolsky, T.; Tucker, C.M.; Chan, L.Y.; Boselli, M.; Dunham, M.J.; et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 2007, 317, 916–924.
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 2010, 188, 369–381.
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.
- Schukken, K.M.; Sheltzer, J. Extensive protein dosage compensation in aneuploid human cancers. Genome Res 2022, 32, 1–31.
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Grönroos, E.; Endesfelder, D.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov 2014, 4, 175–185.
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905.
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Cai, H.; Kumar, N.; Bagheri, H.C.; von Mering, C.; Robinson, M.D.; Baudis, M. Chromothripsis-like patterns are recurring but heterogeneously distributed features in a survey of 22,347 cancer genome screens. BMC Genomics 2014, 15, 1–13.
- Cortés-Ciriano, I.; Lee, J.J.K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet 2020, 52, 331–341.
- Voronina, N.; Wong, J.K.L.; Hübschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; et al. The landscape of chromothripsis across adult cancer types. Nat Commun 2020, 11, 1–13.
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; et al. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184.
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat Genet 2019, 51, 705–715.
- Hassold T; Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet 2001, 2, 280–291.
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962.
- Sack, L.M.; Davoli, T.; Li, M.Z.; Li, Y.; Xu, Q.; Naxerova, K.; et al. Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell 2018, 173, 499–514.
- Norppa, H.; Falck, G.C.M. What do human micronuclei contain? Mutagenesis 2003, 18, 221–233.
- Catalán, J.; Falck, G.C.M.; Norppa, H. The X chromosome frequently lags behind in female lymphocyte anaphase. Am J Hum Genet 2000, 66, 687–691.
- Falck, G.C.; Catala, J.; Norppa, H. Nature of anaphase laggards and micronuclei in female cytokinesis-blocked lymphocytes. Mutagenesis 2002, 17, 111–117.
- Soto, M.; García-Santisteban, I.; Krenning, L.; Medema, R.H.; Raaijmakers, J.A. Chromosomes trapped in micronuclei are liable to segregation errors. J Cell Sci 2018, 131, 1–8.
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell 2013, 154, 47–60.
- Agustinus, A.S.; Raviram, R.; Dameracharla, B.; Luebeck, J.; Stransky, S.; Scipioni, L.; et al. Epigenetic dysregulation from chromosomal transit in micronuclei. BioRxiv 2022.
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E. V.; Pan, Y.; et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58.
- MacKenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; et al. CGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465.
- Payne, A.C.; Chiang, Z.D.; Reginato, P.L.; Mangiameli, S.M.; Murray, E.M.; Yao, C.C.; et al. In situ genome sequencing resolves DNA sequence and structure in intact biological samples. Science 2021, 371.
- Worrall, J.T.; Tamura, N.; Mazzagatti, A.; Shaikh, N.; van Lingen, T.; Bakker, B.; et al. Non-random Mis-segregation of Human Chromosomes. Cell Rep 2018, 23, 3366–3380.
- Torosantucci, L.; De Santis Puzzonia, M.; Cenciarelli, C.; Rens, W.; Degrassi, F. Aneuploidy in mitosis of PtK1 cells is generated by random loss and nondisjunction of individual chromosomes. J Cell Sci 2009, 122, 3455–3461.
- Drpic, D.; Almeida, A.C.; Aguiar, P.; Renda, F.; Damas, J.; Lewin, H.A.; et al. Chromosome Segregation Is Biased by Kinetochore Size. Curr Biol 2018, 28, 1344–1356.
- Klaasen, S.J.; Truong, M.A.; Jaarsveld, R.H.; Koprivec, I.; Štimac, V.; Vries, S.G. De; et al. Nuclear chromosome locations dictate segregation error frequencies. Nature 2022, 607, 604–609.
- Tovini, L.; McClelland, S.E. Impaired CENP-E function renders large chromosomes more vulnerable to congression failure. Biomolecules 2019, 9, 1–15.
- Shaikh, N.; Mazzagatti, A.; Bakker, B.; Spierings, D.C.J.E.; Wardenaar, R.; Muliaditan, D.; et al. Replication Stress Generates Multiple Distinct Classes of Copy Number Alterations. BioRxiv Mol Biol 2019.
- Balajee, A.S.; Bertucci, A.; Taveras, M.; Brenner, D.J. Multicolour FISH analysis of ionising radiation induced micronucleus formation in human lymphocytes. Mutagenesis 2014, 29, 447–455.
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; et al. DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function. Dev Cell 2015, 33, 314–327.
- Dumont, M.; Gamba, R.; Gestraud, P.; Klaasen, S.; Worrall, J.T.; De Vries, S.G.; et al. Human chromosome‐specific aneuploidy is influenced by DNA‐dependent centromeric features. EMBO J 2020, 39, 1–21.
- Spence, J.M.; Mills, W.; Mann, K.; Huxley, C.; Farr, C.J. Increased missegregation and chromosome loss with decreasing chromosome size in vertebrate cells. Chromosoma 2006, 115, 60–74.
- Bochtler, T.; Kartal-Kaess, M.; Granzow, M.; Hielscher, T.; R. Cosenza, M.; Herold-Mende, C.; et al. Micronucleus formation in human cancer cells is biased by chromosome size. Genes Chromosom Cancer 2019, 58, 392–395.
- Klein, A.; Zang, K.D.; Steudel, W.I.; Urbschat, S. Different mechanisms of mitotic instability in cancer cell lines. Int J Oncol 2006, 29, 1389–1396.
- Bell, A.D.; Mello, C.J.; Nemesh, J.; Brumbaugh, S.A.; Wysoker, A.; McCarroll, S.A. Insights into variation in meiosis from 31,228 human sperm genomes. Nature 2020, 583, 259–264.
- Ioannou, D.; Fortun, J.; Tempest, H.G. Meiotic nondisjunction and sperm aneuploidy in humans. Reproduction 2019, 157, 15–31.
- Uroz, L.; Templado, C. Meiotic non-disjunction mechanisms in human fertile males. Hum Reprod 2012, 27, 1518–1524.
- Kuliev, A.; Zlatopolsky, Z.; Kirillova, I.; Spivakova, J.; Cieslak Janzen, J. Meiosis errors in over 20,000 oocytes studied in the practice of preimplantation aneuploidy testing. Reprod Biomed Online 2011, 22, 2–8.
- Gruhn, J.R.; Zielinska, A.P.; Shukla, V.; Blanshard, R.; Newnham, L.J.; Vogel, I.; et al. Chromosome errors in human eggs shape natural fertility over reproductive life span. Science 2019, 365, 1466–1469.
- Hassold, T.; Merrill, M.; Adkins, K.; Freeman, S.; Sherman, S. Recombination and maternal age-dependent nondisjunction: Molecular studies of trisomy 16. Am J Hum Genet 1995, 57, 867–874.
- Fragouli, E.; Wells, D.; Thornhill, A.; Serhal, P.; Faed, M.J.W.; Harper, J.C.; et al. Comparative genomic hybridization analysis of human oocytes and polar bodies. Hum Reprod 2006, 21, 2319–2328.
- Rodriguez-Purata, J.; Lee, J.; Whitehouse, M.; Moschini, R.M.; Knopman, J.; Duke, M.; et al. Embryo selection versus natural selection: How do outcomes of comprehensive chromosome screening of blastocysts compare with the analysis of products of conception from early pregnancy loss (dilation and curettage) among an assisted reproductive technolog. Fertil Steril 2015, 104, 1460–1466.
- Gabriel, A.S.; Thornhill, A.R.; Ottolini, C.S.; Gordon, A.; Brown, A.P.C.; Taylor, J.; et al. Array comparative genomic hybridisation on first polar bodies suggests that non-disjunction is not the predominant mechanism leading to aneuploidy in humans. J Med Genet 2011, 48, 433–437.
- Feichtinger, M.; Stopp, T.; Göbl, C.; Feichtinger, E.; Vaccari, E.; Mädel, U.; et al. Increasing live birth rate by preimplantation genetic screening of pooled polar bodies using array comparative genomic hybridization. PLoS One 2015, 10, 1–13.
- Handyside, A.H.; Montag, M.; Magli, M.C.; Repping, S.; Harper, J.; Schmutzler, A.; et al. Multiple meiotic errors caused by predivision of chromatids in women of advanced maternal age undergoing in vitro fertilisation. Eur J Hum Genet 2012, 20, 742–747.
- Haarhuis, J.H.I.; Elbatsh, A.M.O.; Rowland, B.D. Cohesin and its regulation: On the logic of X-shaped chromosomes. Dev Cell 2014, 31, 7–18.
- Binyam, M.; Scheffler, K.; Schuh, M. Assembly and Positioning of the Oocyte Meiotic Spindle. Annu Rev Cell Dev Biol 2018, 34, 381–403.
- Sacristan, C.; Kops, G.J.P.L. Joined at the hip: Kinetochores, microtubules, and spindle assembly checkpoint signaling. Trends Cell Biol 2015, 25, 21–28.
- Kops, G.J.P.L.; Saurin, A.T.; Meraldi, P. Finding the middle ground : how kinetochores power chromosome congression. Cell Mol Life Sci 2010, 67, 2145–2161.
- Vukušic, K.; Tolic, I.M. Polar Chromosomes — Challenges of a Risky Path. Cells 2022, 11, 1–27.
- Barisic, M.; Aguiar, P.; Geley, S.; Maiato, H. Kinetochore motors drive congression of peripheral polar chromosomes by overcoming random arm-ejection forces. Nat Cell Biol 2014, 16, 1249–1256.
- Roos, U.-P. Light and Electron Microscopy of Rat Kangaroo Cells in Mitosis III. Patterns of Chromosome Behavior during Prometaphase. Chromosoma 1976, 54, 363–385.
- Itoh, G.; Ikeda, M.; Iemura, K.; Amin, M.A.; Kuriyama, S.; Tanaka, M.; et al. Lateral attachment of kinetochores to microtubules is enriched in prometaphase rosette and facilitates chromosome alignment and bi-orientation establishment. Sci Rep 2018, 8, 1–18.
- Pachis, S.T.; Kops, G.J.P.L. Leader of the SAC: Molecular mechanisms of Mps1/TTK regulation in mitosis. Open Biol 2018, 8, 1–10.
- London, N.; Biggins, S. Signalling dynamics in the spindle checkpoint response. Nat Rev Mol Cell Biol 2014, 15, 735–747.
- Etemad, B.; Kuijt, T.E.F.; Kops, G.J.P.L. Kinetochore-microtubule attachment is sufficient to satisfy the human spindle assembly checkpoint. Nat Commun 2015, 6, 1–8.
- Tauchman, E.C.; Boehm, F.J.; DeLuca, J.G. Stable kinetochore-microtubule attachment is sufficient to silence the spindle assembly checkpoint in human cells. Nat Commun 2015, 6, 1–9.
- Lampson, M.A.; Renduchitala, K.; Khodjakov, A.; Kapoor, T.M. Correcting improper chromosomes-spindle attachments during cell division. Nat Cell Biol 2004, 6, 232–237.
- Liu, D.; Vader, G.; Vromans, M.J.M.; Lampson, M.A.; Lens, S.M.A. Sensing Chromosome Bi-Orientation Kinase from Kinetochore Substrates. Science 2009, 323, 1350–1353.
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The Chromosomal Passenger Complex (CPC): From Easy Rider to the Godfather of Mitosis. Nat Rev Mol Cell Biol 2012, 13, 789–803.
- Hindriksen, S.; Lens, S.M.A.; Hadders, M.A. The ins and outs of Aurora B inner centromere localization. Front Cell Dev Biol 2017, 5, 1–21.
- Ohkura, H. Meiosis: An overview of key differences from mitosis. Cold Spring Harb Perspect Biol 2015, 7, 1–15.
- Bolcun-Filas, E.; Handel, M.A. Meiosis: The chromosomal foundation of reproduction. Biol Reprod 2018, 99, 112–126.
- Hengeveld, R.C.C.; Vromans, M.J.M.; Vleugel, M.; Hadders, M.A.; Lens, S.M.A. Inner centromere localization of the CPC maintains centromere cohesion and allows mitotic checkpoint silencing. Nat Commun 2017, 8, 1–12.
- Solomon, D.A.; Kim, T.; Diaz-Martinez, L.A.; Fair, J.; Elkahloun, A.G.; Harris, B.T.; et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 2011, 333, 1039–1043.
- Magidson, V.; Paul, R.; Yang, N.; Ault, J.G.; O’Connell, C.B.; Tikhonenko, I.; et al. Adaptive changes in the kinetochore architecture facilitate proper spindle assembly. Nat Cell Biol 2015, 17, 1134–1144.
- Cherry, L.M.; Johnston, D.A. Size variation in kinetochores of human chromosomes. Hum Genet 1987, 75, 155–158.
- Catalán, J.; Autio, K.; Kuosma, E.; Norppa, H. Age-dependent inclusion of sex chromosomes in lymphocyte micronuclei of man. Am J Hum Genet 1998, 63, 1464–1472.
- Guttenbach, M.; Schakowski, R.; Schmid, M. Aneuploidy and ageing: sex chromosome exclusion into micronuclei. Hum Genet 1994, 94, 295–298.
- Nath, J.; Tucker, J.D.; Hando, J.C. Y Chromosome aneuploidy, micronuclei, kinetochores and aging in men. Chromosoma 1995, 103, 725–731.
- Guttenbach, M.; Koschorz, B.; Bernthaler, U.; Grimm, T.; Schmid, M. Sex chromosome loss and aging: In situ hybridization studies on human interphase nuclei. Am J Hum Genet 1995, 57, 1143–1150.
- Macedo, J.C.; Vaz, S.; Bakker, B.; Ribeiro, R.; Bakker, P.L.; Escandell, J.M.; et al. FoxM1 repression during human aging leads to mitotic decline and aneuploidy-driven full senescence. Nat Commun 2018, 9, 1–17.
- Wang, I.-C.; Chen, Y.-J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; et al. Forkhead Box M1 Regulates the Transcriptional Network of Genes Essential for Mitotic Progression and Genes Encoding the SCF (Skp2-Cks1) Ubiquitin Ligase. Mol Cell Biol 2005, 25, 10875–10894.
- Lee, S.H.; Itkin-Ansari, P.; Levine, F. CENP-A, a protein required for chromosome segregation in mitosis, declines with age in islet but not exocrine cells. Aging (Albany NY) 2010, 2, 785–790.
- McGregor, M.; Hariharan, N.; Joyo, A.Y.; Margolis, R.L.; Sussman, M.A. CENP-A is essential for cardiac progenitor cell proliferation. Cell Cycle 2014, 13, 739–748.
- Kumar, R.; Nagpal, G.; Kumar, V.; Usmani, S.S.; Agrawal, P.; Raghava, G.P.S. HumCFS: A database of fragile sites in human chromosomes. BMC Genomics 2019, 19, 1–8.
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496.
- Bartkova, J.; Horejsi, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870.
- Webster, A.; Schuh, M. Mechanisms of Aneuploidy in Human Eggs. Trends Cell Biol 2017, 27, 55–68.
- Laurie, D.A.; Hulten, M.A. Further studies on bivalent chiasma frequency in human males with normal karyotypes. Ann Hum Genet 1985, 49, 189–201.
- Falek, A.; Chiarelli, B. Meiotic chromosomes of man. Am J Phys Anthropol 1943, 28, 351–354.
- Zelazowski, M.J.; Sandoval, M.; Paniker, L.; Hamilton, H.M.; Han, J.; Gribbell, M.A.; et al. Age-Dependent Alterations in Meiotic Recombination Cause Chromosome Segregation Errors in Spermatocytes. Cell 2017, 171, 601–614.
- Lu, S.; Zong, C.; Fan, W.; Yang, M.; Li, J.; Chapman, A.R.; et al. Probing Meiotic Recombination and Aneuploidy of Single Sperm Cells by Whole-Genome Sequencing. Science 2012, 338, 1627–1631.
- Wang, S.; Hassold, T.; Hunt, P.; White, M.A.; Zickler, D.; Kleckner, N.; et al. Inefficient Crossover Maturation Underlies Elevated Aneuploidy in Human Female Meiosis. Cell 2017, 168, 977–989.
- Sakakibara, Y.; Hashimoto, S.; Nakaoka, Y.; Kouznetsova, A.; Höög, C.; Kitajima, T.S. Bivalent separation into univalents precedes age-related meiosis i errors in oocytes. Nat Commun 2015, 6, 1–8.
- Duncan, F.E.; Hornick, J.E.; Lampson, M.A.; Schultz, R.M.; Shea, L.D.; Woodruff, T.K. Chromosome cohesion decreases in human eggs with advanced maternal age. Aging Cell 2012, 11, 1121–1124.
- Revenkova, E.; Herrmann, K.; Adelfalk, C.; Jessberger, R. Oocyte cohesin expression restricted to predictyate stages provides full fertility and prevents aneuploidy. Curr Biol 2010, 20, 1529–1533.
- Burkhardt, S.; Borsos, M.; Szydlowska, A.; Godwin, J.; Williams, S.A.; Cohen, P.E.; et al. Chromosome Cohesion Established by Rec8-Cohesin in Fetal Oocytes is Maintained without Detectable Turnover in Oocytes Arrested for Months in Mice. Curr Biol 2016, 26, 678–685.
- Zielinska, A.P.; Holubcova, Z.; Blayney, M.; Elder, K.; Schuh, M. Sister kinetochore splitting and precocious disintegration of bivalents could explain the maternal age effect. Elife 2015, 4, 1–19.
- Wang, S.; Liu, Y.; Shang, Y.; Zhai, B.; Yang, X.; Kleckner, N.; et al. Crossover Interference, Crossover Maturation, and Human Aneuploidy. BioEssays 2019, 41, 1–13.
- Capalbo, A.; Hoffmann, E.R.; Cimadomo, D.; Ubaldi, F.M.; Rienzi, L. Human female meiosis revised: New insights into the mechanisms of chromosome segregation and aneuploidies from advanced genomics and time-lapse imaging. Hum Reprod Update 2017, 23, 706–722.
- Bolzer, A.; Kreth, G.; Solovei, I.; Koehler, D.; Saracoglu, K.; Fauth, C.; et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol 2005, 3, 0826–0842.
- Magidson, V.; O’Connell, C.B.; Lončarek, J.; Paul, R.; Mogilner, A.; Khodjakov, A. The spatial arrangement of chromosomes during prometaphase facilitates spindle assembly. Cell 2011, 146, 555–567.
- Cimini, D.; Cameron, L.A.; Salmon, E.D. Anaphase Spindle Mechanics Prevent Mis-Segregation of Merotelically Oriented Chromosomes. Curr Biol 2004, 14, 2149–2155.
- Maiato, H.; Gomes, A.M.; Sousa, F.; Barisic, M. Mechanisms of chromosome congression during mitosis. Biology (Basel) 2017, 6, 1–56.
- Vázquez-Diez, C.; Paim, L.M.G.; FitzHarris, G. Cell-Size-Independent Spindle Checkpoint Failure Underlies Chromosome Segregation Error in Mouse Embryos. Curr Biol 2019, 29, 865–873.
- Cavazza, T.; Takeda, Y.; Politi, A.Z.; Aushev, M.; Aldag, P.; Baker, C.; et al. Parental genome unification is highly error-prone in mammalian embryos. Cell 2021, 184, 2860-2877.e22.
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Andrew, D.; Sanchez-vega, F.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2019, 33, 721–735.
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat Genet 2016, 48, 238–244.
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim Biophys Acta 2017, 1867, 151–161.
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015, 47, 209–216.
- Rutledge, S.D.; Douglas, T.A.; Nicholson, J.M.; Vila-Casadesús, M.; Kantzler, C.L.; Wangsa, D.; et al. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci Rep 2016, 6, 1–12.
- Replogle, J.M.; Zhou, W.; Amaro, A.E.; McFarland, J.M.; Villalobos-Ortiz, M.; Ryan, J.; et al. Aneuploidy increases resistance to chemotherapeutics by antagonizing cell division. Proc Natl Acad Sci 2020, 117, 30566–30576.
- Ben-David, U.; Amon, A. Context is everything: aneuploidy in cancer. Nat Rev Genet 2020, 21, 44–62.
- van Jaarsveld, R.H.; Kops, G.J.P.L. Difference Makers : Chromosomal Instability versus Aneuploidy in Cancer. Trends in Cancer 2016, 2, 561–571.
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018, 173, 581–594.
- Araujo, A.; Bentley, P.; Baum, B. Modelling the role of aneuploidy in tumour evolution. Alife 2010, 488–495.
- Valind, A.; Jin, Y.; Gisselsson, D. Elevated Tolerance to Aneuploidy in Cancer Cells: Estimating the Fitness Effects of Chromosome Number Alterations by In Silico Modelling of Somatic Genome Evolution. PLoS One 2013, 8, 1–17.
- Savva, G.M.; Walker, K.; Morris, J.K. The maternal age-specific live birth prevalence of trisomies 13 and 18 compared to trisomy 21 (Down syndrome). Prenat Diagn 2010, 30, 57–64.
- Bolton, H.; Graham, S.J.L.; Van Der Aa, N.; Kumar, P.; Theunis, K.; Fernandez Gallardo, E.; et al. Mouse model of chromosome mosaicism reveals lineage-specific depletion of aneuploid cells and normal developmental potential. Nat Commun 2016, 7, 1–12.
- Bakhoum, S.F.; Genovese, G.; Compton, D.A. Deviant Kinetochore Microtubule Dynamics Underlie Chromosomal Instability. Curr Biol 2009, 19, 1937–1942.
- Bakhoum, S.F.; Thompson, S.L.; Manning, A.L.; Compton, D.A. Genome stability is ensured by temporal control of kinetochore- microtubule dynamics. Nat Cell Biol 2009, 11, 27–35.
- Pihan, G.A.; Wallace, J.; Zhou, Y.; Doxsey, S.J. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res 2003, 63, 1398–1404.
- Chan, J.Y. A Clinical Overview of Centrosome Amplification in Human Cancers. Int J Biol Sci 2011, 7, 1122–1144.
- Neil J. Ganem, S.A.G. and D.P. A Mechanism Linking Extra Centrosomes to Chromosomal Instability. Physiol Behav 2011, 176, 139–148.
- Kolano, A.; Brunet, S.; Silk, A.D.; Cleveland, D.W.; Verlhac, M.H. Error-prone mammalian female meiosis from silencing the spindle assembly checkpoint without normal interkinetochore tension. Proc Natl Acad Sci 2012, 109, 1858–1867.
- Kyogoku, H.; Kitajima, T.S. Large Cytoplasm Is Linked to the Error-Prone Nature of Oocytes. Dev Cell 2017, 41, 287–298.
- Holubcová, Z.; Blayney, M.; Elder, K.; Schuh, M. Error-prone chromosome-mediated spindle assembly favors chromosome segregation defects in human oocytes. Science 2015, 348, 1143–1147.