Within the 2,5-dioxopiperazine-containing natural products generated by “head-to-tail” cyclization of peptides, those derived from tryptophan allow further structural diversification due to the rich chemical reactivity of the indole heterocycle, which can generate tetracyclic fragments of hexahydropyrrolo[2,3-b]indole or pyrrolidinoindoline skeleton fused to the 2,5-dioxopiperazine. Even more complex are the dimeric bispyrrolidinoindoline epi(poly)thiodioxopiperazines (BPI-ETPs), since they feature transannular (poly)sulfide bridges connecting C3 and C6 of their 2,5-dioxopiperazine rings. Homo- and heterodimers composed of diastereomeric epi(poly)thiodioxopiperazines increase the complexity of the family.
- bispyrrolidinoindoline epi(poly)thiodioxopiperazine alkaloids
- isolation
- structural elucidation
1. Introduction
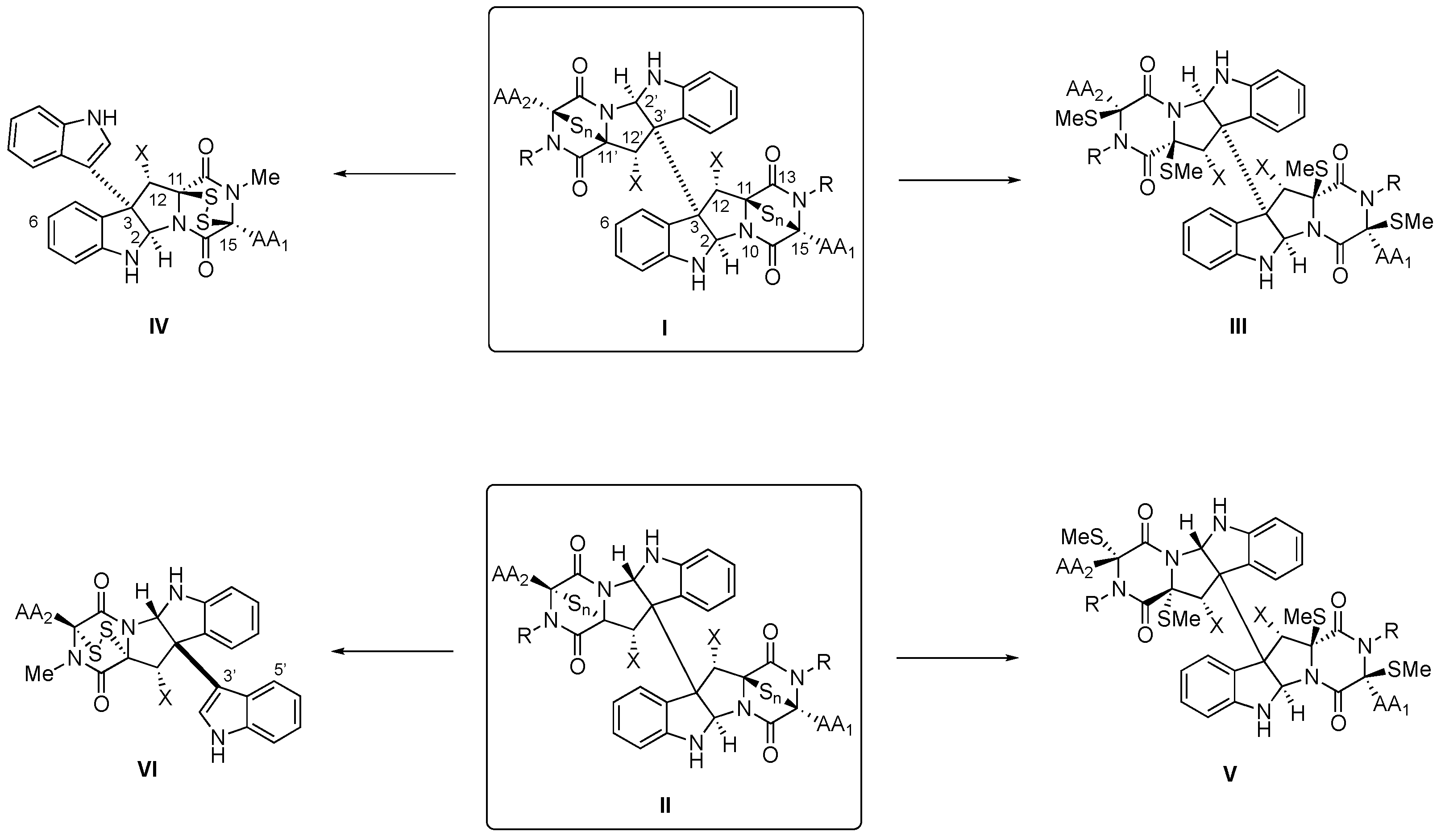
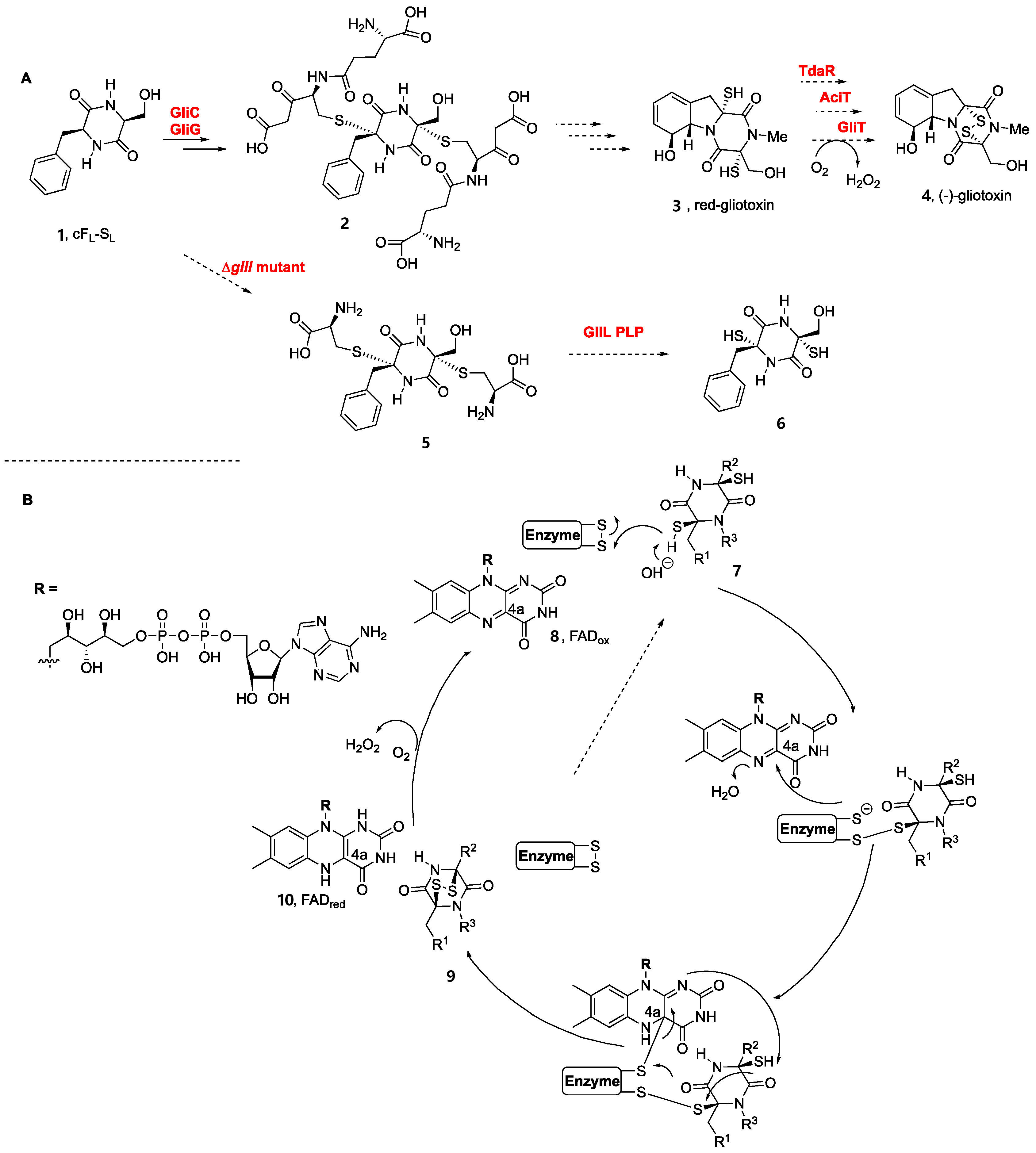
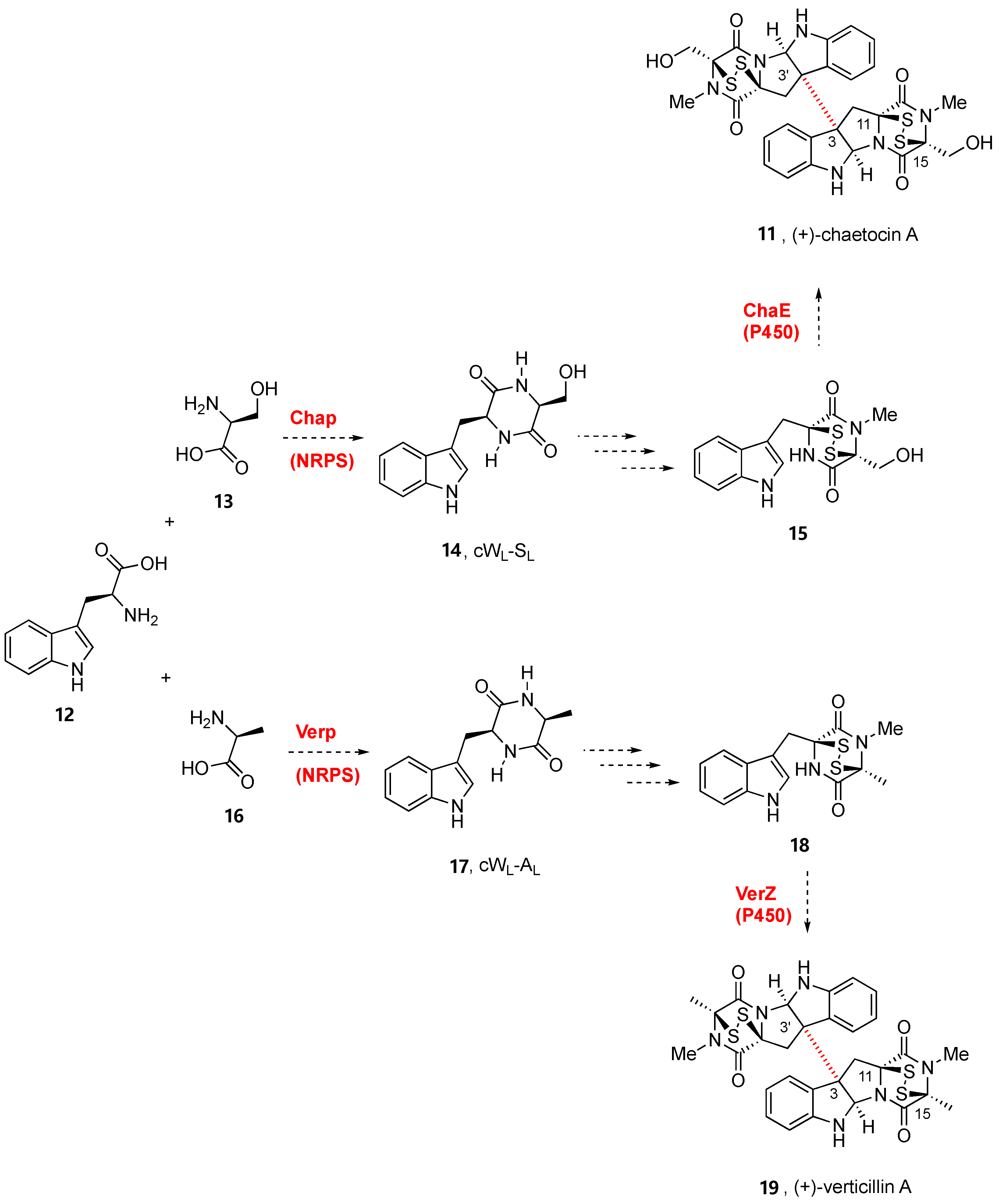
2. Biogenesis of BPI-ETPs
This entry is adapted from the peer-reviewed paper 10.3390/molecules27217585
References
- Wang, X.; Li, Y.; Zhang, X.; Lai, D.; Zhou, L. Structural Diversity and Biological Activities of the Cyclodipeptides from Fungi. Molecules 2017, 22, 2026.
- Bojarska, J.; Mieczkowski, A.; Ziora, Z.M.; Skwarczynski, M.; Toth, I.; Shalash, A.O.; Parang, K.; El-Mowafi, S.A.; Mohammed, E.H.M.; Elnagdy, S.; et al. Cyclic Dipeptides: The Biological and Structural Landscape with Special Focus on the Anti-Cancer Proline-Based Scaffold. Biomolecules 2021, 11, 1515.
- Ma, Z.; Zhou, A.; Xia, C. Strategies for total synthesis of bispyrrolidinoindoline alkaloids. Nat. Prod. Rep. 2022, 39, 1015–1044.
- Barrow, C.J.; Cai, P.; Snyder, J.K.; Sedlock, D.M.; Sun, H.H.; Cooper, R. WIN 64821, a new competitive antagonist to substance P, isolated from an Aspergillus species: Structure determination and solution conformation. J. Org. Chem. 1993, 58, 6016–6021.
- Borthwick, A.D. 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products. Chem. Rev. 2012, 112, 3641–3716.
- Ciarkowski, J. CNDO/2 quantum-mechanical calculations of the conformational flexibility of the diketopiperazine skeleton. Biopolymers 1984, 23, 397–407.
- Liskamp, R.M.J.; Rijkers, D.T.S.; Kruijtzer, J.A.W.; Kemmink, J. Peptides and Proteins as a Continuing Exciting Source of Inspiration for Peptidomimetics. ChemBioChem 2011, 12, 1626–1653.
- Bai, W.-J.; Wang, X. Appreciation of symmetry in natural product synthesis. Nat. Prod. Rep. 2017, 34, 1345–1358.
- Ma, Y.-M.; Liang, X.-A.; Kong, Y.; Jia, B. Structural Diversity and Biological Activities of Indole Diketopiperazine Alkaloids from Fungi. J. Agric. Food Chem. 2016, 64, 6659–6671.
- Gardiner, D.M.; Waring, P.; Howlett, B.J. The epipolythiodioxopiperazine (ETP) class of fungal toxins: Distribution, mode of action, functions and biosynthesis. Microbiology 2005, 151, 1021–1032.
- Jiang, C.-S.; Müller, W.E.G.; Schröder, H.C.; Guo, Y.-W. Disulfide- and Multisulfide-Containing Metabolites from Marine Organisms. Chem. Rev. 2011, 112, 2179–2207.
- Welch, T.R.; Williams, R.M. Epidithiodioxopiperazines. Occurrence, synthesis and biogenesis. Nat. Prod. Rep. 2014, 31, 1376–1404.
- Gomes, N.G.M.; Pereira, R.B.; Andrade, P.B.; Valentão, P. Double the Chemistry, Double the Fun: Structural Diversity and Biological Activity of Marine-Derived Diketopiperazine Dimers. Mar. Drugs 2019, 17, 551.
- Zhang, Q.; Li, H.Q.; Zong, S.C.; Gao, J.M.; Zhang, A.L. Chemical and Bioactive Diversities of the Genus Chaetomium Secondary Metabolites. Mini-Rev. Med. Chem. 2012, 12, 127–148.
- Ishikawa, Y.; Morimoto, K.; Hamasaki, T. Flavoglaucin, a metabolite of Eurotium chevalieri, its antioxidation and synergism with tocopherol. J. Am. Oil Chem. Soc. 1984, 61, 1864–1868.
- Slack, G.J.; Puniani, E.; Frisvad, J.C.; Samson, R.A.; Miller, J.D. Secondary metabolites from Eurotium species, Aspergillus calidoustus and A. insuetus common in Canadian homes with a review of their chemistry and biological activities. Mycol. Res. 2009, 113, 480–490.
- Scott, T.A.; Piel, J. The hidden enzymology of bacterial natural product biosynthesis. Nat. Rev. Chem. 2019, 3, 404–425.
- Giessen, T.W.; Marahiel, M.A. Rational and combinatorial tailoring of bioactive cyclic dipeptides. Front. Microbiol. 2015, 6, 785.
- Hauser, D.; Weber, H.P.; Sigg, H.P. Isolierung und Strukturaufklärung von Chaetocin. Helv. Chim. Acta 1970, 53, 1061–1073.
- Katagiri, K.; Sato, K.; Hakayama, S.; Matsushima, T.; Minato, H. Verticillin A, a new antibiotic from Verticillium sp. J. Antibiot. 1970, 23, 420–422.
- Weindling, R.; Emerson, O.H. The isolation of a toxic substance from the culture filtrate of Trichoderma. Phytopathology 1936, 26, 1068–1070.
- Bell, M.R.; Johnson, J.R.; Wildi, B.S.; Woodward, R.B. THE STRUCTURE OF GLIOTOXIN. J. Am. Chem. Soc. 1958, 80, 1001.
- Walsh, C.T. Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep. 2015, 33, 127–135.
- Walsh, C.T.; Moore, B.S. Enzymatic Cascade Reactions in Biosynthesis. Angew. Chem. Int. Ed. 2018, 58, 6846–6879.
- Gondry, M.; Sauguet, L.; Belin, P.; Thai, R.; Amouroux, R.; Tellier, C.; Tuphile, K.; Jacquet, M.; Braud, S.; Courçon, M.; et al. Cyclodipeptide synthases are a family of tRNA-dependent peptide bond–forming enzymes. Nat. Chem. Biol. 2009, 5, 414–420.
- Gondry, M.; Jacques, I.B.; Thai, R.; Babin, M.; Canu, N.; Seguin, J.; Belin, P.; Pernodet, J.-L.; Moutiez, M. A Comprehensive Overview of the Cyclodipeptide Synthase Family Enriched with the Characterization of 32 New Enzymes. Front. Microbiol. 2018, 9, 46.
- Canu, N.; Moutiez, M.; Belin, P.; Gondry, M. Cyclodipeptide synthases: A promising biotechnological tool for the synthesis of diverse 2,5-diketopiperazines. Nat. Prod. Rep. 2019, 37, 312–321.
- Shende, V.V.; Khatri, Y.; Newmister, S.A.; Sanders, J.N.; Lindovska, P.; Yu, F.; Doyon, T.J.; Kim, J.; Houk, K.N.; Movassaghi, M.; et al. Structure and Function of NzeB, a Versatile C–C and C–N Bond-Forming Diketopiperazine Dimerase. J. Am. Chem. Soc. 2020, 142, 17413–17424.
- Yao, T.; Liu, J.; Liu, Z.; Li, T.; Li, H.; Che, Q.; Zhu, T.; Li, D.; Gu, Q.; Li, W. Genome mining of cyclodipeptide synthases unravels unusual tRNA-dependent diketopiperazine-terpene biosynthetic machinery. Nature Commun. 2018, 9, 4091.
- Canu, N.; Belin, P.; Thai, R.; Correia, I.; Lequin, O.; Seguin, J.; Moutiez, M.; Gondry, M. Incorporation of Non-canonical Amino Acids into 2,5-Diketopiperazines by Cyclodipeptide Synthases. Angew. Chem. Int. Ed. 2018, 57, 3118–3122.
- Borgman, P.; Lopez, R.D.; Lane, A.L. The expanding spectrum of diketopiperazine natural product biosynthetic pathways containing cyclodipeptide synthases. Org. Biomol. Chem. 2019, 17, 2305–2314.
- García-Domínguez, P.; Areal, A.; Alvarez, R.; de Lera, A.R. Chemical synthesis in competition with global genome mining and heterologous expression for the preparation of dimeric tryptophan-derived 2,5-dioxopiperazines. Nat. Prod. Rep. 2022, 39, 1172–1225.
- Zhang, X.; Guo, J.; Cheng, F.; Li, S. Cytochrome P450 enzymes in fungal natural product biosynthesis. Nat. Prod. Rep. 2021, 38, 1072–1099.
- Liu, J.; Liu, A.; Hu, Y. Enzymatic dimerization in the biosynthetic pathway of microbial natural products. Nat. Prod. Rep. 2021, 38, 1469–1505.
- Liu, H.; Fan, J.; Zhang, P.; Hu, Y.; Liu, X.; Li, S.-M.; Yin, W.-B. New insights into the disulfide bond formation enzymes in epidithiodiketopiperazine alkaloids. Chem. Sci. 2021, 12, 4132–4138.
- Patteson, J.B.; Cai, W.; Johnson, R.A.; Maria, K.C.S.; Li, B. Identification of the Biosynthetic Pathway for the Antibiotic Bicyclomycin. Biochemistry 2017, 57, 61–65.
- Meng, S.; Han, W.; Zhao, J.; Jian, X.; Pan, H.; Tang, G. A Six-Oxidase Cascade for Tandem C−H Bond Activation Revealed by Reconstitution of Bicyclomycin Biosynthesis. Angew. Chem. Int. Ed. 2017, 57, 719–723.
- Giessen, T.W.; von Tesmar, A.M.; Marahiel, M.A. A tRNA-Dependent Two-Enzyme Pathway for the Generation of Singly and Doubly Methylated Ditryptophan 2,5-Diketopiperazines. Biochemistry 2013, 52, 4274–4283.
- Yu, H.; Xie, X.; Li, S.-M. Coupling of Guanine with cyclo-l-Trp-l-Trp Mediated by a Cytochrome P450 Homologue from Streptomyces purpureus. Org. Lett. 2018, 20, 4921–4925.
- Shi, J.; Xu, X.; Zhao, E.J.; Zhang, B.; Li, W.; Zhao, Y.; Jiao, R.H.; Tan, R.X.; Ge, H.M. Genome Mining and Enzymatic Total Biosynthesis of Purincyclamide. Org. Lett. 2019, 21, 6825–6829.
- Yu, H.; Xie, X.; Li, S.-M. Coupling of cyclo-L-Trp-L-Trp with Hypoxanthine Increases the Structure Diversity of Guanitrypmycins. Org. Lett. 2019, 21, 9104–9108.
- Liu, J.; Xie, X.; Li, S.-M. Guanitrypmycin Biosynthetic Pathways Imply Cytochrome P450 Mediated Regio- and Stereospecific Guaninyl-Transfer Reactions. Angew. Chem. Int. Ed. 2019, 58, 11534–11540.
- Yet, L. Priviledged Structures in Drug Discovery: Medicinal Chemistry and Synthesis; John Wiley & Sons, Inc.: Hoboken, NY, USA, 2018.
- Cook, K.M.; Hilton, S.T.; Mecinović, J.; Motherwell, W.B.; Figg, W.D.; Schofield, C.J. Epidithiodiketopiperazines Block the Interaction between Hypoxia-inducible Factor-1α (HIF-1α) and p300 by a Zinc Ejection Mechanism. J. Biol. Chem. 2009, 284, 26831–26838.
- Iwasa, E.; Hamashima, Y.; Sodeoka, M. Epipolithiodiketopiperazines Alkaloids: Total Synthesis and Biological Activities. Isr. J. Chem. 2011, 51, 420–433.
- Jiang, C.-S.; Wang, L.; Guo, Y.-W. Epipolythiodioxopiperazines from Fungi: Chemistry and Bioactivities. Recent Advances in Medicinal Chemistry; Atta-ur-Rahman, Choudhary, M.I., Perry, G., Eds.; Bentham eBooks: Soest, The Netherlands, 2015; Volume 2, pp. 76–106.
- Wang, N.; Saidhareddy, P.; Jiang, X. Construction of sulfur-containing moieties in the total synthesis of natural products. Nat. Prod. Rep. 2019, 37, 246–275.
- Hai, Y.; Wei, M.-Y.; Wang, C.-Y.; Gu, Y.-C.; Shao, C.-L. The intriguing chemistry and biology of sulfur-containing natural products from marine microorganisms (1987–2020). Mar. Life Sci. Technol. 2021, 3, 488–518.
- Sun, C.; Tian, W.; Lin, Z.; Qu, X. Biosynthesis of pyrroloindoline-containing natural products. Nat. Prod. Rep. 2022, 39, 1721–1765.
- Dunbar, K.L.; Scharf, D.H.; Litomska, A.; Hertweck, C. Enzymatic Carbon–Sulfur Bond Formation in Natural Product Biosynthesis. Chem. Rev. 2017, 117, 5521–5577.
- Scharf, D.H.; Groll, M.; Habel, A.; Heinekamp, T.; Hertweck, C.; Brakhage, A.A.; Huber, E.M. Flavoenzyme-Catalyzed Formation of Disulfide Bonds in Natural Products. Angew. Chem. Int. Ed. 2014, 53, 2221–2224.
- Harken, L.; Li, S.-M. Modifications of diketopiperazines assembled by cyclodipeptide synthases with cytochrome P450 enzymes. App. Microbiol. Biotechnol. 2021, 105, 2277–2285.
- Scherlach, K.; Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 2021, 12, 1–12.
- Pedras, M.S.C.; Séguin-Swartz, G.; Abrams, S.R. Minor phytotoxins from the blackleg fungus Phoma lingam. Phytochemistry 1990, 29, 777–782.
- Cramer, R.A.; Gamcsik, M.P.; Brooking, R.M.; Najvar, L.K.; Kirkpatrick, W.R.; Patterson, T.F.; Balibar, C.J.; Graybill, J.R.; Perfect, J.R.; Abraham, S.N.; et al. Disruption of a Nonribosomal Peptide Synthetase in Aspergillus fumigatus Eliminates Gliotoxin Production. Eukaryotic Cell 2006, 5, 972–980.
- Scharf, D.H.; Remme, N.; Habel, A.; Chankhamjon, P.; Scherlach, K.; Heinekamp, T.; Hortschansky, P.; Brakhage, A.A.; Hertweck, C. A Dedicated Glutathione S-Transferase Mediates Carbon–Sulfur Bond Formation in Gliotoxin Biosynthesis. J. Am. Chem. Soc. 2011, 133, 12322–12325.
- Davis, C.; Carberry, S.; Schrettl, M.; Singh, I.; Stephens, J.C.; Barry, S.M.; Kavanagh, K.; Challis, G.L.; Brougham, D.; Doyle, S. The Role of Glutathione S-Transferase GliG in Gliotoxin Biosynthesis in Aspergillus fumigatus. Chem. Biol. 2011, 18, 542–552.
- Scharf, D.H.; Chankhamjon, P.; Scherlach, K.; Heinekamp, T.; Willing, K.; Brakhage, A.A.; Hertweck, C. Epidithiodiketopiperazine Biosynthesis: A Four-Enzyme Cascade Converts Glutathione Conjugates into Transannular Disulfide Bridges. Angew. Chem. Int. Ed. 2013, 52, 11092–11095.
- Gerken, T.; Walsh, C.T. Cloning and Sequencing of the Chaetocin Biosynthetic Gene Cluster. ChemBioChem 2013, 14, 2256–2258.
- Wang, Y.; Hu, P.; Pan, Y.; Zhu, Y.; Liu, X.; Che, Y.; Liu, G. Identification and characterization of the verticillin biosynthetic gene cluster in Clonostachys rogersoniana. Fungal Genet. Biol. 2017, 103, 25–33.
- Guo, Z.; Hao, T.; Wang, Y.; Pan, Y.; Ren, F.; Liu, X.; Che, Y.; Liu, G. VerZ, a Zn(II)2Cys6 DNA-binding protein, regulates the biosynthesis of verticillin in Clonostachys rogersoniana. Microbiology 2017, 163, 1654–1663.
- Zhu, S.; Ren, F.; Guo, Z.; Liu, J.; Liu, X.; Liu, G.; Che, Y. Rogersonins A and B, Imidazolone N-Oxide-Incorporating Indole Alkaloids from a verG Disruption Mutant of Clonostachys rogersoniana. J. Nat. Prod. 2019, 82, 462–468.
- Wang, Y.; Ren, J.; Li, H.; Pan, Y.; Liu, X.; Che, Y.; Liu, G. The disruption of verM activates the production of gliocladiosin A and B in Clonostachys rogersoniana. Org. Biomol. Chem. 2019, 17, 6782–6785.