Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Organic
|
Medicine, Research & Experimental
Alkaloids containing indoles—indole alkaloids—are among the most significant alkaloid subsets, with more than 4100 different compounds.
- indole
- natural products
- cytotoxic
1. Introduction
Despite the decreasing interest of modern pharmaceuticals in pursuing natural products as leads for new medicine [1], natural products and their derivatives still represent a significant fraction of approved drugs, with more than 22 compounds in the WHO list of essential medicine being sourced exclusively from flowering plants [2]. Furthermore, approximately 40% of all available medicine is either a natural product or a semi-synthetic derivative [3][4]. This ubiquity of natural products in medicine can be explained by their inherent nature as secondary metabolites. These compounds have been fine-tuned over millennia, acquiring biological functions to boost their host’s survivability [5]. Thus, these compounds can regulate endogenous defense mechanisms and interactions with other organisms, explaining their potency as antiviral, antibacterial, and antitumor agents [6]. Natural products also represent desired targets in synthesis due to their structural complexity or potent bioactivity.
Additionally, the pursuit of synthesizing natural products has led to novel reaction methodologies, catalyst design, and natural product-inspired synthetic analogs [7][8]. However, so far, only 5–15% of terrestrial plant species have been investigated as potential sources of therapeutic agents. Similarly, despite accounting for 90% of all-natural diversity, less than 1% of the microbial domain has been explored [1]. This minimal investigation of natural products’ sources (plants, insects, and microorganisms) represents a significant opportunity for chemists to scrutinize these sources and further discover nature’s offerings [9].
Due to their immense structural diversity, natural products are generally grouped into four broad categories: phenolics (phenylpropanoids), polyketides, terpenoids, and alkaloids [10][11]. Alkaloids are nitrogen-containing secondary metabolites, including important therapeutic agents such as hyoscyamine, quinine, or emetine [12][13]. Additionally, alkaloids are further divided into classes based on which nitrogen-containing moiety they possess; tropane, isoquinoline, imidazole, piperidine, and pyrrolizidine alkaloids are some examples [14].
Alkaloids containing indoles—indole alkaloids—are among the most significant alkaloid subsets, with more than 4100 different compounds [15]. Commercially available drugs having the indole moiety are ajmaline (antiarrhythmic agent), physostigmine (used to treat anticholinergic poisoning), and vincristine (antitumor agent) [16][17][18]. Similarly, many controlled substances such as ibogaine, psilocybin, and LSD (all powerful psychedelics) feature an indole moiety, demonstrating the group’s ubiquity in bioactive molecules [19].
2. Synthesis of Indole Alkaloids
2.1. Total Synthesis of Indole Alkaloids
(±)-Conolidine (269), a potent nonopioid analgesic, was synthesized in six steps without any nonstrategic redox manipulations. It was isolated from the stem bark of Tabernaemontana divaricate in 2004 [20], and its first total synthesis was carried out in 2011 with a nine-step synthetic route in an 18% overall yield [21]. The latest total synthesis attempts primarily utilized gold(I)-catalyzed Conia-ene and Pictet–Spengler reactions, producing 269 in six steps in overall 19% yield (Figure 16). They also used DFT calculations to develop a successful scheme [22].
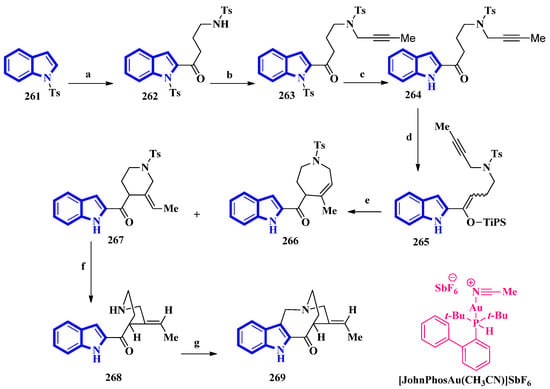
Figure 16. The total synthesis of (±)-conolidine 269. (a) n-BuLi, THF, −78 °C, 5 min, then rt, 1 h, then N-tosylpyrrolidonein THF, −30 °C to −15 °C, 4 h, 48%; (b) K2CO3, 1-bromo-2-butyne, CH3CN, 80 °C, 94%; (c) TBAF, CH3CN 35 °C, 15 h, 93%; (d) TiPSOTf, 2,6-lutidine, 35 °C, 5 h, 97%, E:Z = 8:92; (e) [JohnPhosAu(CH3CN)]SbF6, H2O, toluene, 60 °C, 2 h, 15% for 266, 73% for 267; (f) Sodium naphthalenide, THF, −78 °C, 86%; (g) (CH2O)n, TFA, CH3CN, reflux, 2 h, 82%.
For the total synthesis of compound 269, compound 261 was treated with n-butyllithium and dropwise addition to N-tosylpyrrolidone affording 262 that underwent a nucleophilic reaction with 1-bromo-2-butyne to give compound 263. Next, compound 263 was treated with TBAF to remove the N-1-tosyl group producing compound 264. Afterward, 264 was added to TiPSOTf in 2,6-lutidine, affording a pair of stereoisomers of 265 (E:Z = 8:92). Its gold-catalyzed reaction using [JohnPhosAu(CH3CN)]SbF6 produced compounds 266 and 267. The latter was exposed to sodium naphthalenide to deprotect its N-2 tosyl group affording 268. Lastly, a Pictet–Spengler reaction using TFA and paraformaldehyde in acetonitrile was used to convert 268 into 269 [22].
Psammocindoles A and B (212 and 213) significantly increased adiponectin production. Thus, to further investigate their pharmacology and assign the absolute configuration of compound 212 at C-2′, the two alkaloids and their congener, psammocindole C (214), were synthesized. In addition, enantiomers of psammocindole D (286), and four N-lactam analogs, isopsammocindoles A–D (287 to 290), were produced. The synthetic scheme was four steps long, and the overall yields were 23%, 21%, and 21% for compounds 212, 213, and 214, respectively (Figure 17). As a result, the R-configuration of compound 212 at C-2′ was assigned [23].
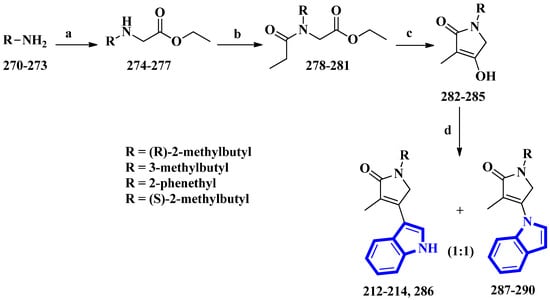
Figure 17. The total synthesis of psammocindoles (A–D) 212–214, 286, and their isomers 287–290. (a) Ethyl bromoacetate, DCM, Et3N, rt, 2 h, 90–93%; (b) Propionyl chloride, Et3N, 0 °C to rt, 1.5 h, 94–95%; (c) NaH, THF, reflux, 12 h, 83–86%; (d) Indole, BF3-Et2O, 4 Å MS, PhCl, 100 °C, 1.5 h, 27–31%.
(R)-2-Methylbutan-1-amine was prepared in an 88% enantiomeric excess using a five-step synthetic scheme, while the other amines were obtained commercially. The amines (270 to 273) were reacted with ethyl bromoacetate, affording compounds 274 to 277. Subsequently, further condensation with propionyl chloride produced compounds 278 to 281, which were used to prepare the N-alkyl-α,β-unsaturated γ-lactams (282 to 285) by an intramolecular Claisen condensation. Lastly, condensation between the lactams (at C-3) and the indole’s nitrogen afforded compounds 212 to 214 and 286 to 290, with approximately 30% yields for all analogs [23].
To confirm the structure of amakusamine (233), it was synthesized by the same research group that first isolated it. Upon initial failure of a direct debromination of 5,6-methylenedioxyindole, they used an alternative reaction scheme containing three steps and obtained an overall yield of 56% (Figure 18). The synthetic method also enabled the synthesis of 15 synthetic derivatives of compound 233 (294 to 308) for an SAR analysis (Figure 19) [24]. The synthesis involved the reaction of 6-nitropiperonal (291) with NBS in concentrated H2SO4, producing 292. A Henry reaction (using Al2O3 as a base and nitromethane) followed by dehydration using acetic anhydride transformed 292 into 293. However, since the latter was crystalline and hard to dissolve in many solvents, its crude mixture was reduced directly by an excess of iron in acetic acid, affording compound 233 [24].
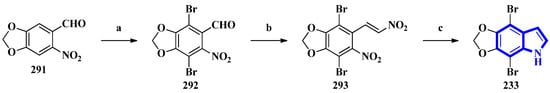
Figure 18. The total synthesis of amakusamine 233. (a) NBS, conc. H2SO4, 63%; (b) (1) Al2O3, CH3NO2 and (2) Ac2O; (c) Iron in AcOH, (last two steps) 89%.
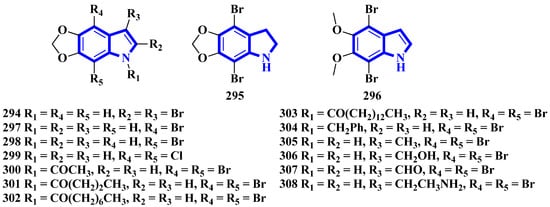
Figure 19. Amakusamine derivatives synthesized 294–308.
The first total synthesis of griseofamine B (314) and its three stereoisomers, 16-epi-griseofamine B, ent-griseofamine B, and 11-epi-griseofamine B (317, 321, and 323), was carried out in 2022. Compound 314 is an indole-tetramic acid alkaloid isolated from the Penicillium griseofulvum fungus in 2018 [25]. In the total synthesis, 4-bromo tryptophan methyl ester hydrochloride was used as the starting reagent, with its l-enantiomer being used to synthesize compounds 314 and 317. d-Enantiomer 4-bromo tryptophan methyl ester hydrochloride was used to synthesize compounds 321 and 323. All four compounds were obtained in five steps with yields of 18%, 5%, 19%, and 5%, respectively (Figure 20) [26]. Next, compound 309 was reacted with Boc2O, Et3N, and DMAP under reflux conditions in DCM to afford 310 in 90% yield. A Heck–Mizoroki reaction of 310 with 2-methyl-3-buten-2-ol in the presence of PdCl2(PPh3)4, Ag2CO3, and Et3N in 1,4-dioxane produced compound 311 in 67% yield. Afterward, refluxing 311 with PdCl2(CH3CN)2 in CH3CN generated a pair of diastereomers 312 and 315. TMSOTf and 2,6-lutidine in DCM were used to remove the Boc groups and gave compounds 313 and 316. A tandem acylation/Lacey–Dieckmann cyclization of 313 and 316 with diketene was performed to afford 314 and 317, respectively. Additionally, using the same synthetic scheme with compound 318 as the starting reagent afforded compounds 321 and 323 [26].
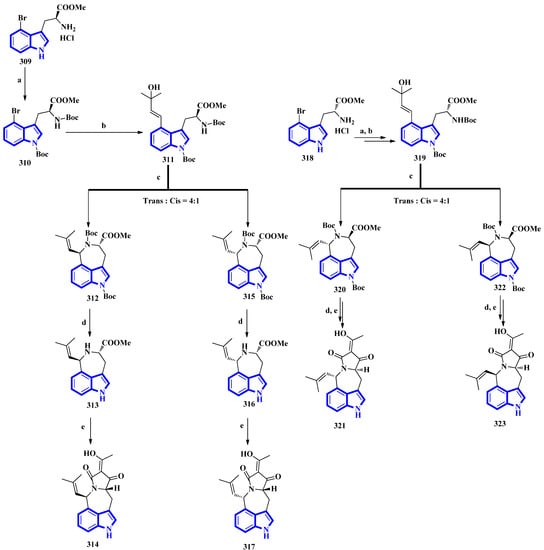
Figure 20. The total synthesis of griseofamine B (314) and its isomers 317, 321, and 323. (a) Boc2O, Et3N, DMAP, DCM, reflux, 2 h, 90%; (b) 2-methyl-3-buten-2-ol, PdCl2(PPh3)4, Ag2CO3, Et3N, 1,4-dioxane, 100 °C, 6 h; 67% (c) PdCl2(CH3CN)2, CH3CN, reflux, 2 h; E:Z = 56–58%:15–16% (d) TMSOTf, 2,6-lutidine, DCM, rt, overnight, 87–88%; (e) diketene, Et3N, DCM, rt, overnight, 62–63%.
2.2. Synthesis of Indole Derivatives
As β-carboline derivatives generally display only moderate cytotoxicity, new synthetic derivatives were designed to contain a hydroxycinnamic acid moiety inferring HDAC-inhibitory properties that provide synergy and improve their antiproliferative effects [27]. These derivatives (347 to 352) differed only in the number of carbons connecting the β-carboline with the hydroxycinnamic acid motif and in the substituents on the phenyl ring in the hydroxycinnamic acid moiety (Figure 21).
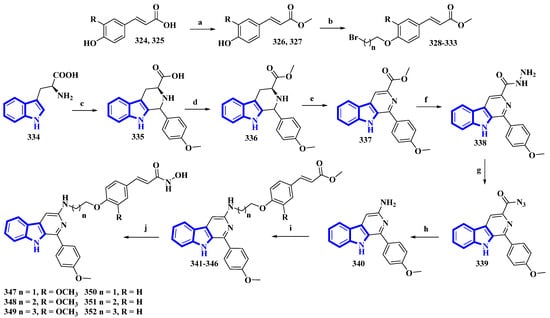
Figure 21. Synthesis of hydroxamic derivatives 347–352. (a) SOCl2, CH3OH, 0 °C, 1 h, and then 65 °C, 4 h; (b) ω-Dibromoalkane, K2CO3, CH3CN, reflux, 4 h, 66–75%; (c) 4-methoxybenzaldehyde, AcOH, reflux, 4 h; (d) Thionyl chloride, CH3OH, 0 °C, 1 h, and then 65 °C, 6 h; (e) KMnO4, DMF, rt, 5 h; (f) Hydrazine monohydrate, CH3OH, 50 °C, 6 h; (g) NaNO2, HCl; (h) AcOH, H2O, 50 °C, 6 h; (i) 328–333, K2CO3, CH3CN, reflux, 12 h; (j) NH2OK, CH3OH, rt, 8–12 h, 46–56%.
(E)-Ferulic acid (324) and p-coumaric acid (325) were esterified using SOCl2 in CH3OH, yielding compounds 326 and 327. These compounds were then treated with ω-dibromoalkanes (1,2-dibromoethane, 1,2-dibromopropane, and 1,2-dibromobutane) in the presence of K2CO3 to produce compounds 328 to 333. Using a Pictet–Spengler reaction, l-tryptophan (334) was transformed into 335 with 4-methoxybenzaldehyde. Compound 335 was then esterified using SOCl2 in CH3OH, giving compound 336, which was oxidized using KMnO4 in DMF, affording compound 337. It was then reacted with hydrazine monohydrate to give compound 338 and was converted into 339 using NaNO2. Next, compound 339 underwent a Curtis rearrangement to produce compound 340. Lastly, compounds 328 to 333 were reacted with 340, yielding residues 341 to 346, which were then treated with NH2OK to produce derivatives 347 to 352.
Neopeltolide, a marine natural product, was isolated from a sponge in the neopeltidae family. It is a highly potent antitumor agent (IC50 < 1 nm) and strongly inhibits cytochrome bc1, an essential component of the mitochondrial respiratory chain [28]. Compounds (404 to 424 and 438 to 443) were synthesized, replacing its macrolactone ring with an indole having potential as a fungicide. The derivatives were synthesized in two series. The first series (404 to 424) contained an ester linkage between the oxazole and indole heterocycles (Figure 22). The second series (313 to 318), guided through the bioactivities of the compounds in the first series (Table 2) and the docking studies of leads, replaced the linkage with an amide group and contained only fluorine or methoxy substituents (Figure 22) [29]. The reaction of prop-2-yn-1-amine (353) with NaHCO3 in 1,4-dioxane affords 354. Carboxylation of 354 using n-butyllithium in THF and a CO2 atmosphere produced 355 that was reduced to 356 using Lindlar’s catalyst. Compound 356 reacted with l-serine methyl ester hydrochloride, N-methylmorpholine, and isobutyl chloroformate to produce 357. Compound 357 was reacted with DAST, DBU, and BrCCl3 at a low temperature to yield 358, and LiOH hydrolyzed it to give 359. Separately, methyl indole-4-carboxylate (360) was treated with N-chlorosuccinimide in acidic conditions, producing 361. It was then reacted with substituted benzyl chlorides and NaH in THF, yielding 362 to 382, which were reduced by DIBAL-H, giving 383 to 403. Lastly, 359 was reacted with each of the 21 intermediates to give derivatives 404 to 424. The second derivatives were synthesized by reacting 4-nitroindole (425) with substituted benzyl bromides and NaH in anhydrous DMF, yielding 426 to 431. They were reduced using iron and NH4Cl in EtOH and water, affording 432 to 437. Another time, 359 reacted with each of the six derivatives, producing derivatives of 438 to 443 [29].
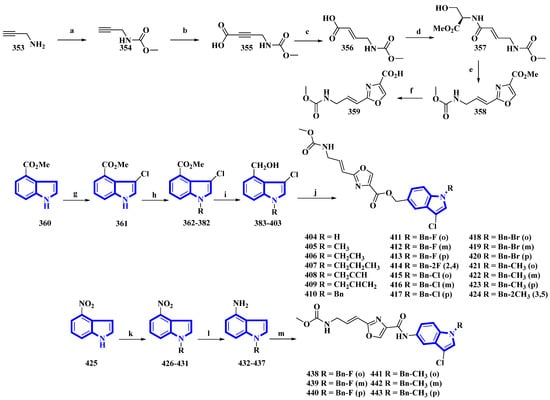
Figure 22. Synthesis of neopeltolide derivatives 404 to 424 and 438 to 443. (a) NaHCO3, 1,4-dioxane, rt, 95%; (b) n-BuLi, CO2, THF, −78 °C, 84%; (c) H2, Lindlar’s catalyst, EtOAc, 91%; (d) l-Serine methyl ester hydrochloride, i-BuOCOCl, N-CH3-morpholine, THF, 75%; (e) (1) DAST, DCM, −78 °C and (2) BrCCl3, DBU, −20 °C, 62%; (f) LiOH, THF/H2O, 88%; (g) NCS, HCl, THF, 82%; (h) Substituted benzyl chlorides, NaH, DMF, rt, quantitative; (i) DIBAL-H, THF, −78 °C, 57–75%; (j) 359, EDCI, HOBt, Et3N, DMF, 43–71%; (k) substituted benzyl bromides, NaH, DMF, rt, quantitative; (l) Fe, NH4Cl, H2O, EtOH, reflux, 53–67%; (m) 359, EDCI, HOBt, DMF, 49–70%.
Celastrol (444) is a friedelane-type triterpenoid isolated from Tripterygium wilfordii and possesses immunosuppressive properties [30]. However, it has toxic properties. Therefore, its synthetic derivatives were prepared to obtain lower cytotoxicity. Ten celastrol derivatives (456 to 465) with indole substituents were synthesized that differed only in the substituents attached to the indole group (Figure 23) [31]. In the synthesis, compound 444 was converted into 445 via a nucleophilic reaction using propargyl bromide and NaHCO3 in DMF at room temperature. Compound 445 was transformed using a Friedel–Crafts reaction using substituted indoles and FeCl3·6H2O in DCM, giving compounds 446 to 455. These compounds were acetylated using DMAP and Ac2O immediately without purification, yielding derivatives 456 to 465.
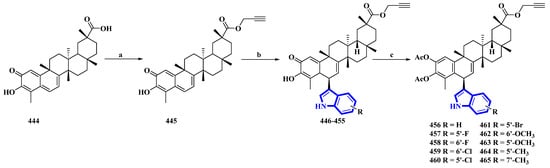
Figure 23. Synthesis of celastrol derivatives 456 to 465. (a) NaHCO3, propargyl bromide, DMF, rt, 78%; (b) substituted indoles, FeCl3.6H2O, DCM; (c) Ac2O, DMAP, DCM, 37–54%.
Phidianidine A is a marine natural product isolated from Phidiana militaris, a mollusk. The natural product and its synthetic analogs possess both cytotoxic and immunosuppressive activities. However, the antifouling properties of its derivatives were not previously explored despite its resemblance with other potent antifouling MNPs. Therefore, 10 synthetic derivatives (479 to 488) having primary amines, guanidines, and a quaternary ammonium compound were produced (Figure 24) [32]. Initially, the diamines 466 to 469 (propan-1,3-diamine, butan-1,4-diamine, and pentan-1,5-diamine) and 6-aminohexanol (474) were protected using Boc2O. Then, compound 474 was converted into compound 475 using methane sulfonyl chloride and methylamine. Separately, 6-bromoindole (476) was converted into derivative 477 using oxalyl chloride. It was then reduced to compound 478 using hydrazine and sodium methoxide. The derivative 478 was then reacted with the diamines 470 to 473 and 475 and deprotected using TFA, affording the primary amines 479 to 483. Additionally, compounds 479 to 481 and 483 were reacted with DIPEA and 489 to yield guanidine derivatives 484 to 487. Furthermore, compound 479 was reacted with NaBH3CN in formaldehyde and acetic acid, producing quaternary ammonium derivative 488 [32].
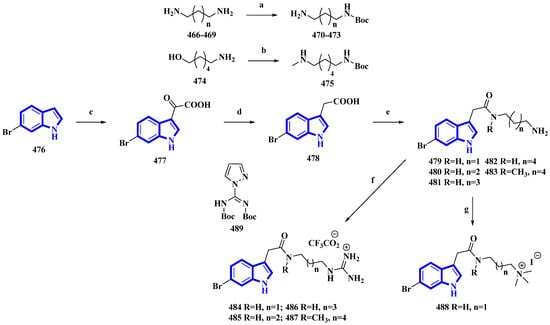
Figure 24. Synthesis of phidianidine A derivatives 479 to 488. (a) Boc2O, rt, overnight, 93–95%; (b) (1) Boc2O, Et3N, rt, 2 h, (2) MsCl, Et3N, 0 °C, 5 h and (3) CH3NH2, 60 °C, 1 h, 58%; (c) oxalyl chloride, 30 min, 77%; (d) (1) Hydrazine, 80 °C, MW, 15 min and (2) NaOCH3, 80 °C, MW, 15 min, 80%; (e) (1) 470–473, 475, DIPEA, HATU, 1.5 h and (2) TFA, DCM, 4 h, 8–73%; (f) (1) 479–481, 483, 489, DIPEA, 3 h and (2) TFA, DCM, 2 h, 24–83%; (g) 479, CH2O, NaBH3CN, AcOH, 21 h and (2) CH3I, 0 °C, 53%.
2.3. Semi-Synthesis of Indole Alkaloids
Fradcarbazole A, a novel staurosporine-type indole alkaloid containing a thiazole group, was isolated from a mutant strain of Streptomyces fradiae and semi-synthesized from staurosporine by the same group [33][34]. To enhance its efficacy as an antitumor agent, 14 derivatives (535 to 548) of fradcarbazole A were synthesized by variation only in the substituents on two indole units (Figure 25) [35]. In the synthesis, 5-fluoro/chloro/bromo/methoxy-tryptamines (489 to 493) were protected using Boc2O to give derivatives 494 to 498 and oxidized using DDQ, producing compounds 499 to 503. TFA-mediated Boc deprotection resulted in molecules 504 to 508. Staurosporine (509) was protected using Boc2O, giving 510, which was halogenated using NCS and NBS (compounds 511 and 512), and deprotected by TFA (compounds 513 and 514). These derivatives and compound 509 were converted to compounds 515 to 517 using TCDI. These compounds were alkylated by CH3I in CH3CN, affording compounds 518 to 520. They were then converted into compounds 521 to 534 via a nucleophilic reaction with 504 to 508. Lastly, intramolecular cyclization reactions of compounds 521 to 534 resulted in 535 to 548 [35].
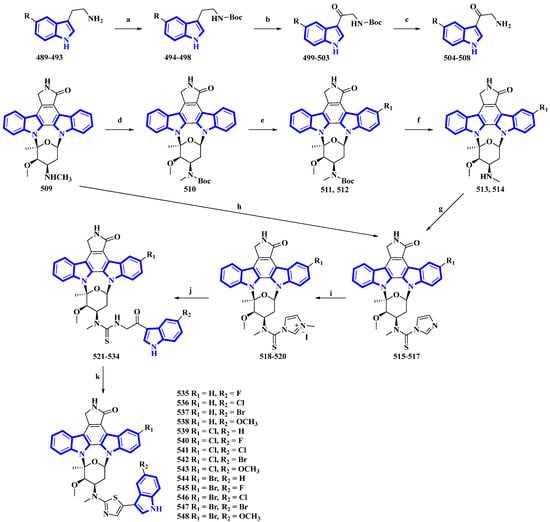
Figure 25. Synthesis of fradcarbazole A derivatives 535 to 548. (a) Boc2O, Et3N, THF, 10 °C, 83–99%; (b) DDQ, THF/H2O 0 °C, 58–81%; (c) TFA, 10 °C, 72–94%; (d) Boc2O, Et3N, THF, 0 °C, 76%; (e) NBS, CH3OH, DCM, 0 °C, 94%, or NCS, CH3OH, DCM, rt, 48%; (f) TFA, DCM, 0 °C, 50–93%; (g) TCDI, Et3N, DCM, rt, 71–88%; (h) TCDI, Et3N, DCM, rt, 82%; (i) CH3I, CH3CN, rt, 68–74%; (j) 504–508, Et3N, DMF, rt, 38–56%; (k) (CF3CO)2O, DCM, EtOH, 0 °C, 47–81%.
(−)-Melodinine K (556), a complex bisindole alkaloid, was semi-synthesized, using (−)-tabersonine as the starting reagent. The aspidosperma–aspidosperma-type alkaloid was isolated from the Melodinus tenuicaudatus plant in 2010. Its cytotoxicity was tested by the same group and was reported to be more potent than cisplatin and vinorelbine in four of the five cancer cell lines [36]. The synthetic route adopted had six steps in its most extended linear sequence (eight steps overall) and afforded compound 556 with a 4% yield (Figure 26) [37]. While planning its synthesis, 556 was divided into two fragments, both of which could be synthesized from (−)-tabersonine (549). The total synthesis of 549 was accomplished by the same group in 2013 [38]. However, this precursor was mainly isolated from the seeds of Voacanga africana (a small tree). It was bio-transformed into compound 550 using T16H yeast and allylated by allyl bromide and K2CO3 in DMF, resulting in 551, the northern fragment of compound 556. Separately, compound 549 was protected using TrocCl, affording 552, which was further converted to 553 by TFA and m-CPBA. Compound 553 was reacted with m-CPBA in DCM, producing its N-oxide analog 554. Compound 554 was treated with TFAA in DCM and coupled with 551 in a Polonovski–Potier reaction, which resulted in 555. Compound 555 was deprotected by treating with Pd(PPh3)4, affording 556 [37].
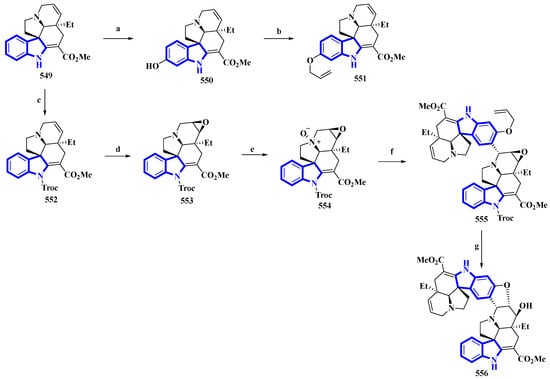
Figure 26. The semi-synthesis of (−)-melodinine K 556. (a) T16H yeast, 64%; (b) Allyl bromide, K2CO3, DMF, 68%; (c) NaH, TrocCl, THF/DMF 0 °C, 92%; (d) TFA then m-CPBA, DCM, −10 °C then rt, 38%; (e) m-CPBA, DCM, 0 °C, 36%; (f) (1) TFAA, DCM, 0 °C to rt and (2) 551, DCM, 78%; (g) (1) Pd(PPh3)4, pyrrolidine, DCM, rt and (2) Zn, KH2PO4, THF, 60 °C, 40%.
5-Methylpsilocybin (561), a novel analog of the psychedelic psilocybin, was produced by enzymatically phosphorylating its synthetic precursor, 5-methylpsilocin (560). The 4-hydroxytryptamine kinase (PsiK) enzyme was purified from the Psilocybe cubensis fungus. On the basis of the amount of 561 isolated, the overall yield was 28% (Figure 27) [39]. 5-Methyl-1H-indol-4-ol (557) was acetylated with acetic anhydride and NaHCO3 in toluene into compound 558. It was then treated with oxalyl chloride, followed by dimethylamine in THF, to result in 559, which, on reduction by LiAlH4, resulted in 560. Compound 560 was incubated with PsiK and ATP for 16 h; the analysis of the mixture by HPLC indicated that 90% of it was successfully converted into 5-methylpsilocybin (561) with an overall isolated yield of only 40% [39].
Table 2. Bioactivities of all novel indole alkaloids discussed in the article. (a) Tested as a mixture; (b) KB/VJ300 cells grown with vincristine (0.1 μM), which did not affect their growth; (c) methicillin-resistant strain; (d) IC90; (e) IC40.
| Compound | Cytotoxicity (GI50) µM [Cell Line] | Ref. |
|---|---|---|
| 1 | 38 [Huh7], 32 [LN229], 19 [HCT116], 16 [MGC803], 16 [A549], 21 [MDA231] | [40] |
| 15 | 1.1 [KB/S], 4.1 [KB/VJ300 b], 4.7 [PC-3], 4.8 [MCF7], 3.6 [MDA-MB-231], 4.3 [HCT 116], 2.2 [HT-29h] | [41] |
| 16 | 0.1 [KB/S], 1.3 [KB/VJ300 b], 7.9 [PC-3], 9.6 LNCaP, 0.6 [MCF7], 1.4 [MDA-MB-231], 1.1 [HCT 116], 0.3 [HT-29h] | [41] |
| 17 | 0.1 [KB/S], 8.6 [KB/VJ300], 1.5 [KB/VJ300 b], 0.7 [PC-3], 0.3 [MCF7], 0.4 [MDA-MB-231], 0.2 [HCT 116], 0.2 [HT-29h] | [41] |
| 18 | 0.2 [KB/S], 2.6 [LNCaP], 3.5 [MCF7], 5.4 [MDA-MB-231], 3.1 [HCT 116], 2.3 [HT-29h] | [41] |
| 24 | 3.6 [L5178Y], 8.7 [A2780b], 40 [J82], 29 [HEK-293] | [42] |
| 25 | 5.3 [L5178Y], 12 [A2780b], 42 [J82], 22 [HEK-293] | [42] |
| 26 | 5.3 [L5178Y], 28 [HEK-293] | [42] |
| 27 | 12 [A2780b], 55 [J82] | [42] |
| 28 | 32 [A2780b], 100 [J82] | [42] |
| 29 | 8.1 [L5178Y], 7.8 [A2780b], 32 [J82], 37 [HEK-293] | [42] |
| 37 | 6.2 [HT-29] | [43] |
| 51–54a | 4.1 × 10−4 [U87], 7.5 × 10−4 [SKOV3], 5.4 [MDA-MB-231 and HCT116] | [44] |
| 55 | 7.3 [HeLa] | [45] |
| 56 | 6.4 [HeLa] | [45] |
| 71 | 0.6 [KB/S], 6.7 [KB/VJ300], 0.2 [KB/VJ300 b], 8.2 [PC-3], 6.2 [LNCaP], 4.5 [MCF7], 4.5 [MDA-MB-231], 0.3 [HT-29], 7 [A549], 7.9 [MRC-5] | [46] |
| 72 | 2.8 [KB/S], 5.9 [KB/VJ300], 6.1 [PC-3], 2 [MCF7], 4.8 [MDA-MB-231], 0.07 [HT-29], 4.2 [HCT 116], 9 [A549], 9.7 [CCD-18Co] | [46] |
| 73 | 1.1 [KB/S], 3.9 [KB/VJ300B], 3.7 [LNCaP], 5.3 [MCF7], 4.8 [MDA-MB-231], 0.07 [HT-29] | [46] |
| 74 | 2.4 [KB/S], 2.9 [KB/VJ300], 3.1 [PC-3], 1.3 [MCF7], 1.4 [MDA-MB-231], 0.03 [HT-29], 2.9 [HCT 116], 6.8 [A549], 4.3 [CCD-18Co] | [46] |
| 75 | 2.5 [KB/S], 5.5 [KB/VJ300], 8.8 [KB/VJ300 b], 7.7 [MCF7], 4.1 [MDA-MB-231], 0.03 [HT-29], 1.5 [HCT 116], 3.9 [A549], 4.1 [CCD-18Co] | [46] |
| 76 | 1.7 [KB/S], 3.6 [KB/VJ300], 4.2 [KB/VJ300 b], 8.5 [MCF7], 0.02 [HT-29], 2.1 [HCT 116], 3 [A549], 5.1 [CCD-18Co] | [46] |
| 117 | 81 [HeLa] | [47] |
| 118 | 42 [HeLa] | [47] |
| 119 | 84 [Huh7], 92 [HeLa] | [47] |
| 123 | 2.45 [A549], 0.96 [Huh7], 0.93 [HeLa] | [47] |
| 145 | 7.5 [SW480] | [48] |
| 157 | 18.7 [HepG2], 28.7 [A-549] | [49] |
| 160 | 17 [U251], 4.5 [U87MG] | [50] |
| 161 | 36 [U251], 43 [U87MG] | [50] |
| 162 | 12 [U251], 2.3 [U87MG] | [50] |
| 164 | 27 [U251], 8.9 [U87MG] | [50] |
| 166 | 36 [U251], 29 [U87MG] | [50] |
| 167 | 19 [U251], 17 [U87MG] | [50] |
| 168 | 12 [U251], 7.4 [U87MG] | [50] |
| 170 | 13 [U251], 11 [U87MG] | [50] |
| 172 | 15 [U251], 19 [U87MG] | [50] |
| 187 | 6.8 [SK-MEL-28], 9.8 [SW480], 6.3 [HepG2], 9.8 [T47D] | [51] |
| 191 | 8.3 [SK-MEL-28], 7.4 [HepG2] | [51] |
| 192 | 6.7 [SK-MEL-28], 7.8 [SW480], 2.5 [HepG2], 8.7 [T47D] | [51] |
| 193 | 9.5 [T47D] | [51] |
| 211 | 5 [KB/S], 7.6 [KB/VJ300], 6.1 [PC-3], 5.9 [MDA-MB-231], 0.2 [HT-29], 4.7 [HCT 116], 2.1 [A549] | [52] |
| 217 | 44.0 [A549], 40.4 [HT-29], 37.3 [HepG2] | [53] |
| 218 | 8.2 [A549], 4.7 [A427], 8.0 [HCT116], 6.7 [HT-29], 6.2 MCF-7, 7.1 [HeLa], 9.1 [HepG2], 44.1 [LO2] | [53] |
| 219 | 13.8 [A427], 38.5 [HCT116], 20.3 [HeLa] | [53] |
| 220 | 18.9 [HeLa], 46.3 [LO2] | [53] |
| 221 | 7.9 [A549], 6.6 [A427], 10.6 [HCT116], 8.2 [HT-29], 7.2 MCF-7, 8.4 [HeLa], 9.6 [HepG2], 35.5 [LO2] | [53] |
| 222 | 9.5 [A549], 4.8 [A427], 12.2 [HCT116], 8.9 [HT-29], 6.5 MCF-7, 8.6 [HeLa], 9.1 [HepG2], 36.3 [LO2] | [53] |
| 223 | 6.9 [A549], 7.5 [A427], 11.2 [HCT116], 8.8 [HT-29], 6.8 MCF-7, 10.1 [HeLa], 10.7 [HepG2], 41.8 [LO2] | [53] |
| 224 | 21.5 [A549], 26.1 [HCT116], 41.5 [HeLa] | [53] |
| 225 | 23.3 [A549], 30.9 [HCT116], 43.8 [HT-29], 44.4 [HepG2] | [53] |
| 226 | 15.9 [A549], 21.2 [HCT116] | [53] |
| 227 | 40.4 [A549], 45.3 [HCT116] | [53] |
| 239 | 1.7 [HeLa], 1.6 [A549], 1.8 [HepG2], 1.5 [SMMC7721] | [54] |
| 242 | 7.9 [HeLa], 7.8 [A549], 8.1 [HepG2], 6.7 [SMMC7721] | [54] |
| 347 | 4 [SMMC-7721], 4.8 [HepG2], 1.8 [Bel7402], 2.8 [Huh7] | [27] |
| 348 | 2.9 [SMMC-7721], 1.4 [HepG2], 1.0 [Bel7402], 1.1 [Huh7] | [27] |
| 349 | 7.1 [SMMC-7721], 5.3 [HepG2], 4.6 [Bel7402], 6.2 [Huh7] | [27] |
| 350 | 6.3 [SMMC-7721], 5.8 [HepG2], 5.2 [Bel7402] | [27] |
| 351 | 4.1 [SMMC-7721], 3.4 [HepG2], 4.0 [Bel7402] | [27] |
| 352 | 9.5 [HepG2], 8.9 [Bel7402] | [27] |
| 535 | 0.51 [MV4-11] | [35] |
| 536 | 0.42 [MV4-11] | [35] |
| 537 | 0.41 [MV4-11] | [35] |
| 538 | 0.36 [MV4-11] | [35] |
| 539 | 0.56 [MV4-11] | [35] |
| 540 | 0.32 [MV4-11] | [35] |
| 541 | 0.59 [MV4-11] | [35] |
| 542 | 0.43 [MV4-11] | [35] |
| 543 | 0.44 [MV4-11] | [35] |
| 544 | 0.36 [MV4-11] | [35] |
| 545 | 0.96 [MV4-11] | [35] |
| 546 | 0.70 [MV4-11] | [35] |
| 547 | 0.39 [MV4-11] | [35] |
| 548 | 0.51 [MV4-11] | [35] |
| Antibacterial (MIC; µg/mL) | ||
| 1 | 25 [B. cereus], 12.5 [S. aureus c] | [40] |
| 2 | 50 [S. aureus c] | [40] |
| 8 | 12.5 [B. cereus], 25 [S. aureusc], 25 [M. lysodeikticus], 25 [B. paratyphosum], 25 [B. subtilis], 25 [E. aerogenes], 25 [S. typhi], 25 [P. vulgaris] | [40] |
| 32 | 4 [B. subtilis], 16 [S. typhimurium], 8 [M. luteus], 8 [M.phlei] | [55] |
| 33 | 32 [B. subtilis], 32 [S. typhimurium], 32 [M. luteus], 32 [M. phlei] | [55] |
| 40 | 8 [X. o. pv. oryzae], 32 [R. solanacearum], 32 [X. o. pv. oryzicola], 128 [P. s. pv. lachrymans] | [56] |
| 42 | 32 [X. o. pv. oryzae] | [56] |
| 44 | 32 [X. o. pv. oryzae], 128 [R. solanacearum], 64 [X. o. pv. oryzicola] | [56] |
| 45 | 64 [X. o. pv. oryzae] | [56] |
| 64 | 25 [H. influenzae ATCC 4] | [57] |
| 65 | 50 [H. influenzae ATCC 4] | [57] |
| 66 | 25 [H. influenzae ATCC 4] | [57] |
| 231 | 25 [M. smegmatis], 25 [M. abscessus], 25 [M. bovis] | [58] |
| Antifungal (MIC; µg/mL) | ||
| 1 | 6.25 [A. fragariae], 25 [C. cassiicola], 25 [A. alternata], 6.25 [B. cinereal Pers], 25 [C. personata], 6.25 [V. dahliaekleb], 25 [S. sclerotiorum] | [40] |
| 2 | 25 [A. fragariae] | [40] |
| 3 | 25 [A. fragariae] | [40] |
| 4 | 50 [A. fragariae] | [40] |
| 5 | 6.25 [A. fragariae] | [40] |
| 6 | 25 [A. fragariae] | [40] |
| 7 | 6.25 [A. fragariae] | [40] |
| 9 | 50 [A. fragariae] | [40] |
| 10 | 25 [A. fragariae] | [40] |
| 229 | 50 [F. oxysporum] | [59] |
| Antiplasmodial (IC50 µM) | ||
| 63 | 6.1 d [P. falciparum] | [60] |
| 110 | 8.7 [P. falciparum FcB1] | [61] |
| 111 | 9.5 [P. falciparum FcB1] | [61] |
| 112 | 2.6 [P. falciparum FcB1] | [61] |
| 113 | 5.2 [P. falciparum FcB1] | [61] |
| 114 | 3.0 [P. falciparum FcB1] | [61] |
| 210 | 1.05 [P. falciparum FcB1] | [62] |
| 234 | 15.1 [P. falciparum 3D7] | [63] |
| Antiviral (EC50 µM) | ||
| 109 | 70 [HSV-2] | [64] |
| 137 | 5.7 [HCV] | [65] |
| 196 | 7.5 [ZIKV] | [66] |
| 197 | 38 [ZIKV] | [66] |
| 198 | 50 [ZIKV] | [66] |
| 249 | 9.4 [H1N1], 6.7 [RSV] | [67] |
| 250 | 3.7 [H1N1], 2.8 [RSV] | [67] |
| Antifouling activity (EC50 µM) | ||
| 486 | 2.2 [A. improvisus] | [32] |
| 487 | 0.7 [A. improvisus] | [32] |
| Inhibition of protein phosphatases (IC50 µM) | ||
| 67 | 14 [PTP1B], 38 [PTPsigma] | [68] |
| 68 | 27 [PTP1B] | [68] |
| 70 | 23 [PTP1B], 35 [TCPTP] | [68] |
| Inhibition of DC secretion of IL-12p40 (% inhibition at 10 µg/mL) | ||
| 177 | 38% | [69] |
| 185 | 36% | [69] |
| Promotion of DC secretion of IL-10 (% promotion at 10 µg/mL) | ||
| 176 | 19% | [69] |
| Promotion of hBM-MSC secretion of adiponectin (EC50 µM) | ||
| 212 | 9.86 | [23] |
| 213 | 6.20 | [23] |
| Inhibition of LPS-induced B-cell proliferation (IC50 µM) | ||
| 237 | 0.38 | [70] |
| 238 | 47.37 | [70] |
| Analgesic activity | ||
| 205 | 64.7% (1 mg/kg) | [71] |
| 206 | 50% (5 mg/kg) | [71] |
| 207 | 67.6% (0.04 mg/kg), 76.1% (0.2 mg/kg) | [71] |
| 208 | 55% (5 mg/kg) | [71] |
| 209 | 53% (5 mg/kg) | [71] |
| 5-HT1A receptor agonist (EC50 µM) | ||
| 83 | 10 | [72] |
| 84 | 2.2 | [72] |
| 86 | 0.1 | [72] |
| 87 | 54 | [72] |
| Inhibition of HDACs (EC50 nM) | ||
| 347 | 32 [HDAC1], 125 [HDAC3], 17 [HDAC6] | [27] |
| 348 | 27 [HDAC1], 148 [HDAC3], 13 [HDAC6] | [27] |
| 349 | 61 [HDAC1] | [27] |
| 350 | 75 [HDAC1] | [27] |
| 351 | 57 [HDAC1] | [27] |
| 352 | 125 [HDAC1] | [27] |
| Inhibition of porcine SCR (EC50 µM) | ||
| 411 | 10 | [29] |
| 438 | 1.34 | [29] |
| 439 | 1.75 | [29] |
| 440 | 1.46 | [29] |
| 442 | 0.70 | [29] |
| Inhibition of acetylcholinesterase (IC50 µM) | ||
| 98 | 10.5 | [73] |
| Inhibition of quorum sensing (MIC µg/well) | ||
| 104 | 32 [C. violaceum CV026] | [74] |
| 105 | 32 [C. violaceum CV026] | [74] |
| Glucose uptake activity in L6 myoblasts at 50 µM (mmol/L) | ||
| 203 | 5 | [75] |
| 204 | 5 | [75] |
| Triglyceride accumulation promotion in 3T3-L1 cells (EC50 µM) | ||
| 126 | 1.03 | [76] |
| Inhibition of MAO-A (IC50 µM) | ||
| 127 | 0.9 [MAO-A] | [77] |
| Inhibition of misc. enzymes (inhibition % at 10 µM) | ||
| 127 | 31 [MAO-B], 21 [BChE] | [77] |
| Inhibition of mushroom tyrosinase (IC50 µM) | ||
| 141 | 33.2 | [78] |
| Inhibition of autophagic flux (EC50 µM) | ||
| 145 | 20.2 | [48] |
| Vasorelaxant activity (EC50 µM) | ||
| 158 | 2.8 | [49] |
| 159 | 2.4 | [49] |
| Potentiation of imipenem activity (4 ug/mL of imipenem; MIC µg/mL) | ||
| 173 | 8–16 [S. aureus c] | [79] |
| 174 | 2–4 [S. aureus c] | [79] |
| Inhibition of RANKL-induced multinuclear osteoclasts (IC50 µM) | ||
| 233 | 10.5 [RAW264] | [24] |
| 294 | 50 e [RAW264] | [24] |
| 295 | 25.6 [RAW264] | [24] |
| 296 | 6.3 [RAW264] | [24] |
| 297 | 16.8 [RAW264] | [24] |
| 298 | 35.4 [RAW264] | [24] |
| 299 | 40.0 [RAW264] | [24] |
| 300 | 11.7 [RAW264] | [24] |
| 301 | 7.9 [RAW264] | [24] |
| 302 | 11.1 [RAW264] | [24] |
| 305 | 8.1 [RAW264] | [24] |
| 306 | 10.0 [RAW264] | [24] |
| 307 | 13.9 [RAW264] | [24] |
| 308 | 5.9 [RAW264] | [24] |
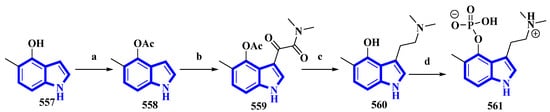
Figure 27. The semi-synthesis of 5-methylpsilocybin 561. (a) Ac2O, NaHCO3, toluene, 97%; (b) (1) oxalyl chloride and (2) (CH3)2NH, THF, 77%; (c) LiAlH4, THF, 84%; (d) PsiK, ATP, MgCl2, H2O, 16 h, 44%.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27217586
References
- Veeresham, C. Natural products derived from plants as a source of drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200.
- John, J.E. Natural products-based drug discovery: Some bottlenecks and considerations. Curr. Sci. 2009, 96, 753–754.
- Eddershaw, P.J.; Beresford, A.P.; Bayliss, M.K. ADME/PK as part of a rational approach to drug discovery. Drug Discov. Today 2000, 5, 409–414.
- Appendino, G.; Fontana, G.; Pollastro, F. Natural Products Drug Discovery. In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 205–236.
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216.
- Mohr, J.T.; Krout, M.R.; Stoltz, B.M. Natural products as inspiration for the development of asymmetric catalysis. Nature 2008, 455, 323–332.
- Grigalunas, M.; Burhop, A.; Christoforow, A.; Waldmann, H. Pseudo-natural products and natural product-inspired methods in chemical biology and drug discovery. Curr. Opin. Chem. Biol. 2020, 56, 111–118.
- Lahlou, M. The success of natural products in drug discovery. Pharmacol. Pharm. 2013, 4, 17–31.
- Li, J.W.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165.
- Hunter, P. Harnessing nature’s wisdom. Turning to nature for inspiration and avoiding her follies. EMBO Rep. 2008, 9, 838–840.
- Hanson, J.R. Natural Products: The Secondary Metabolites; Royal Society of Chemistry: Cambridge, UK, 2003; Volume 17.
- Harden, R.N. Chronic neuropathic pain: Mechanisms, diagnosis, and treatment. Neurologist 2005, 11, 111–122.
- Esu, E.B.; Effa, E.E.; Opie, O.N.; Meremikwu, M.M. Artemether for severe malaria. Cochrane Database Syst. Rev. 2019, 6, CD010678.
- Kaur, R.; Arora, S. Alkaloids-important therapeutic secondary metabolites of plant origin. J. Crit. Rev. 2015, 2, 1–8.
- Seigler, D.S. Plant Secondary Metabolism; Springer: New York, NY, USA, 2001; p. 628.
- Kiesecker, C.; Zitron, E.; Luck, S.; Bloehs, R.; Scholz, E.P.; Kathofer, S.; Thomas, D.; Kreye, V.A.; Katus, H.A.; Schoels, W.; et al. Class Ia anti-arrhythmic drug ajmaline blocks HERG potassium channels: Mode of action. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 370, 423–435.
- Moore, P.W.; Rasimas, J.J.; Donovan, J.W. Physostigmine is the antidote for anticholinergic syndrome. J. Med. Toxicol. 2015, 11, 159–160.
- Avendaño, C.; Menéndez, J.C. (Eds.) Chapter 8—Anticancer drugs targeting tubulin and microtubules. In Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2008; pp. 229–249.
- Koenig, X.; Hilber, K. The anti-addiction drug ibogaine and the heart: A delicate relation. Molecules 2015, 20, 2208–2228.
- Kam, T.S.; Pang, H.S.; Choo, Y.M.; Komiyama, K. Biologically active ibogan and vallesamine derivatives from Tabernaemontana divaricata. Chem. Biodivers. 2004, 1, 646–656.
- Tarselli, M.A.; Raehal, K.M.; Brasher, A.K.; Streicher, J.M.; Groer, C.E.; Cameron, M.D.; Bohn, L.M.; Micalizio, G.C. Synthesis of conolidine, a potent non-opioid analgesic for tonic and persistent pain. Nat. Chem. 2011, 3, 449–453.
- Chen, G.; Wang, C.; Zou, L.; Zhu, J.; Li, Y.; Qi, C. Six-step total synthesis of(+/-)-conolidine. J. Nat. Prod. 2019, 82, 2972–2978.
- Kwon, O.; Ahn, S.; Jeon, J.; Park, I.G.; Won, T.H.; Sim, C.J.; Park, H.; Oh, D.; Oh, K.; Noh, M. Psammocindoles A–C: Isolation, synthesis, and bioactivity of indole-γ-lactams from the sponge Psammocinia vermis. Org. Lett. 2021, 23, 4667–4671.
- Maeyama, Y.; Nakashima, Y.; Kato, H.; Hitora, Y.; Maki, K.; Inada, N.; Murakami, S.; Inazumi, T.; Ise, Y.; Sugimoto, Y.; et al. Amakusamine from a Psammocinia sp. Sponge: Isolation, synthesis, and SAR study on the inhibition of RANKL-induced formation of multinuclear osteoclasts. J. Nat. Prod. 2021, 84, 2738–2743.
- Zang, Y.; Genta-Jouve, G.; Zheng, Y.; Zhang, Q.; Chen, C.; Zhou, Q.; Wang, J.; Zhu, H.; Zhang, Y. Griseofamines A and B: Two indole-tetramic acid alkaloids with 6/5/6/5 and 6/5/7/5 ring systems from Penicillium griseofulvum. Org. Lett. 2018, 20, 2046–2050.
- Sheng, T.; Ma, C.; Zhang, G.; Pan, X.; Liu, Z. Asymmetric total synthesis of griseofamine B and its three stereoisomers. J. Nat. Prod. 2022, 85, 1128–1133.
- Ling, Y.; Li, Y.; Zhu, R.; Qian, J.; Liu, J.; Gao, W.; Meng, C.; Miao, J.; Xiong, B.; Qiu, X.; et al. Hydroxamic acid derivatives of β-carboline/hydroxycinnamic acid hybrids inducing apoptosis and autophagy through the PI3K/Akt/mTOR pathways. J. Nat. Prod. 2019, 82, 1442–1450.
- Wright, A.E.; Botelho, J.C.; Guzman, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J. Nat. Prod. 2007, 70, 412–416.
- Xiong, M.Q.; Chen, T.; Wang, Y.X.; Zhu, X.L.; Yang, G.F. Design and synthesis of potent inhibitors of bc1 complex based on natural product neopeltolide. Bioorg. Med. Chem. Lett. 2020, 30, 127324.
- Venkatesha, S.H.; Moudgil, K.D. Celastrol suppresses experimental autoimmune encephalomyelitis via MAPK/SGK1-regulated mediators of autoimmune pathology. Inflamm. Res. 2019, 68, 285–296.
- He, Q.W.; Feng, J.H.; Hu, X.L.; Long, H.; Huang, X.F.; Jiang, Z.Z.; Zhang, X.Q.; Ye, W.C.; Wang, H. Synthesis and biological evaluation of celastrol derivatives as potential immunosuppressive agents. J. Nat. Prod. 2020, 83, 2578–2586.
- Labriere, C.; Elumalai, V.; Staffansson, J.; Cervin, G.; Le Norcy, T.; Denardou, H.; Rehel, K.; Moodie, L.W.K.; Hellio, C.; Pavia, H.; et al. Phidianidine A and synthetic analogues as naturally inspired marine antifoulants. J. Nat. Prod. 2020, 83, 3413–3423.
- Wang, L.; Mei, X.; Wang, C.; Zhu, W. Biomimetic semi-synthesis of fradcarbazole A and its analogues. Tetrahedron 2015, 71, 7990–7997.
- Fu, P.; Zhuang, Y.; Wang, Y.; Liu, P.; Qi, X.; Gu, K.; Zhang, D.; Zhu, W. New indolocarbazoles from a mutant strain of the marine-derived actinomycete Streptomyces fradiae 007M135. Org. Lett. 2012, 14, 6194–6197.
- Li, M.; Xu, Y.; Zuo, M.; Liu, W.; Wang, L.; Zhu, W. Semisynthetic derivatives of fradcarbazole A and their cytotoxicity against acute myeloid leukemia cell lines. J. Nat. Prod. 2019, 82, 2279–2290.
- Feng, T.; Li, Y.; Wang, Y.Y.; Cai, X.H.; Liu, Y.P.; Luo, X.D. Cytotoxic indole alkaloids from Melodinus tenuicaudatus. J. Nat. Prod. 2010, 73, 1075–1079.
- Walia, M.; Teijaro, C.N.; Gardner, A.; Tran, T.; Kang, J.; Zhao, S.; O’Connor, S.E.; Courdavault, V.; Andrade, R.B. Synthesis of(-)-melodinine K: A case study of efficiency in natural product synthesis. J. Nat. Prod. 2020, 83, 2425–2433.
- Zhao, S.; Andrade, R.B. Domino Michael/Mannich/N-alkylation route to the tetrahydrocarbazole framework of aspidosperma alkaloids: Concise total syntheses of (-)-aspidospermidine, (-)-tabersonine, and (-)-vincadifformine. J. Am. Chem. Soc. 2013, 135, 13334–13337.
- Fricke, J.; Sherwood, A.M.; Halberstadt, A.L.; Kargbo, R.B.; Hoffmeister, D. Chemoenzymatic synthesis of 5-methylpsilocybin: A tryptamine with potential psychedelic activity. J. Nat. Prod. 2021, 84, 1403–1408.
- Xu, L.L.; Hai, P.; Zhang, S.B.; Xiao, J.F.; Gao, Y.; Ma, B.J.; Fu, H.Y.; Chen, Y.M.; Yang, X.L. Prenylated indole diterpene alkaloids from a mine-soil-derived Tolypocladium sp. J. Nat. Prod. 2019, 82, 221–231.
- Sim, D.S.; Navanesan, S.; Sim, K.S.; Gurusamy, S.; Lim, S.H.; Low, Y.Y.; Kam, T.S. Conolodinines A-D, Aspidosperma-Aspidosperma bisindole alkaloids with antiproliferative activity from Tabernaemontana corymbosa. J. Nat. Prod. 2019, 82, 850–858.
- Ariantari, N.P.; Ancheeva, E.; Wang, C.; Mandi, A.; Knedel, T.O.; Kurtan, T.; Chaidir, C.; Muller, W.E.G.; Kassack, M.U.; Janiak, C.; et al. Indole diterpenoids from an endophytic Penicillium sp. J. Nat. Prod. 2019, 82, 1412–1423.
- Wong, S.K.; Wong, S.P.; Sim, K.S.; Lim, S.H.; Low, Y.Y.; Kam, T.S. A cytotoxic indole characterized by incorporation of a unique carbon-nitrogen skeleton and two pentacyclic corynanthean alkaloids incorporating a substituted tetrahydrofuranone ring from Kopsia arborea. J. Nat. Prod. 2019, 82, 1902–1907.
- Kim, M.C.; Cullum, R.; Machado, H.; Smith, A.J.; Yang, I.; Rodvold, J.J.; Fenical, W. Photopiperazines A-D, photosensitive interconverting diketopiperazines with significant and selective activity against U87 glioblastoma cells, from a rare, marine-derived Actinomycete of the family Streptomycetaceae. J. Nat. Prod. 2019, 82, 2262–2267.
- Li, H.; Xu, D.; Sun, W.; Yang, B.; Li, F.; Liu, M.; Wang, J.; Xue, Y.; Hu, Z.; Zhang, Y. HPLC-DAD-directed isolation of linearly fused prenylated indole alkaloids from a soil-derived Aspergillus versicolor. J. Nat. Prod. 2019, 82, 2181–2188.
- Yeap, J.S.; Saad, H.M.; Tan, C.H.; Sim, K.S.; Lim, S.H.; Low, Y.Y.; Kam, T.S. Macroline-sarpagine bisindole alkaloids with antiproliferative activity from Alstonia penangiana. J. Nat. Prod. 2019, 82, 3121–3132.
- Wang, M.H.; Zhang, X.Y.; Tan, X.M.; Niu, S.B.; Sun, B.D.; Yu, M.; Ding, G.; Zou, Z.M. Chetocochliodins A-I, epipoly(thiodioxopiperazines) from Chaetomium cochliodes. J. Nat. Prod. 2020, 83, 805–813.
- Zhang, Y.; Ding, X.; Yuan, Y.X.; Guo, L.L.; Hao, X.J. Cytotoxic monoterpenoid indole alkaloids from Tabernaemontana corymbosa as potent autophagy inhibitors by the attenuation of lysosomal acidification. J. Nat. Prod. 2020, 83, 1432–1439.
- Zhang, J.; Liu, Z.W.; Li, Y.; Wei, C.J.; Xie, J.; Yuan, M.F.; Zhang, D.M.; Ye, W.C.; Zhang, X.Q. Structurally diverse indole alkaloids with vasorelaxant activity from Melodinus hemsleyanus. J. Nat. Prod. 2020, 83, 2313–2319.
- Qin, L.; Yi, W.; Lian, X.Y.; Zhang, Z. Bioactive alkaloids from the Actinomycete Actinoalloteichus sp. ZZ1866. J. Nat. Prod. 2020, 83, 2686–2695.
- Yi, W.F.; Ding, X.; Chen, Y.Z.; Adelakun, T.A.; Zhang, Y.; Hao, X.J. Tabernaesines A-I, cytotoxic Aspidosperma-Aspidosperma-type bisindole alkaloids from Tabernaemontana pachysiphon. J. Nat. Prod. 2020, 83, 3215–3222.
- Tan, C.H.; Yeap, J.S.; Lim, S.H.; Low, Y.Y.; Sim, K.S.; Kam, T.S. The bisindole alkaloids Angustilongines M and A from Alstonia penangiana induce mitochondrial apoptosis and G0/G1 cell cycle arrest in HT-29 cells through promotion of tubulin polymerization. J. Nat. Prod. 2021, 84, 1524–1533.
- Duan, F.F.; Liu, L.; Gao, Y.; Peng, X.G.; Meng, X.G.; Ruan, H.L. -Chaetoglobosins from Pseudeurotium bakeri induce G2/M cell cycle arrest and apoptosis in human cancer cells. J. Nat. Prod. 2021, 84, 1904–1914.
- Guo, X.; Meng, Q.; Liu, J.; Wu, J.; Jia, H.; Liu, D.; Gu, Y.; Liu, J.; Huang, J.; Fan, A.; et al. Sclerotiamides C-H, notoamides from a marine gorgonian-derived fungus with cytotoxic activities. J. Nat. Prod. 2022, 85, 1067–1078.
- Mudalungu, C.M.; von Torne, W.J.; Voigt, K.; Ruckert, C.; Schmitz, S.; Sekurova, O.N.; Zotchev, S.B.; Sussmuth, R.D. Noursamycins, chlorinated cyclohexapeptides identified from molecular networking of Streptomyces noursei NTR-SR4. J. Nat. Prod. 2019, 82, 1478–1486.
- Yan, W.; Zhao, S.S.; Ye, Y.H.; Zhang, Y.Y.; Zhang, Y.; Xu, J.Y.; Yin, S.M.; Tan, R.X. Generation of indoles with agrochemical significance through biotransformation by Chaetomium globosum. J. Nat. Prod. 2019, 82, 2132–2137.
- Bankeu, J.J.K.; Kagho, D.U.K.; Fongang, Y.S.F.; Toghueo, R.M.K.; Mba’ning, B.M.; Feuya, G.R.T.; Fekam, F.B.; Tchouankeu, J.C.; Ngouela, S.A.; Sewald, N.; et al. Constituents from Nauclea latifolia with anti-haemophilus influenzae type b inhibitory activities. J. Nat. Prod. 2019, 82, 2580–2585.
- Fouotsa, H.; Le Pogam, P.; Mkounga, P.; Lannang, A.M.; Bernadat, G.; Vanheuverzwijn, J.; Zhou, Z.; Leblanc, K.; Rharrabti, S.; Nkengfack, A.E.; et al. Voatriafricanines A and B, trimeric Vobasine-Aspidosperma-Aspidosperma alkaloids from Voacanga africana. J. Nat. Prod. 2021, 84, 2755–2761.
- Jiang, C.X.; Yu, B.; Miao, Y.M.; Ren, H.; Xu, Q.; Zhao, C.; Tian, L.L.; Yu, Z.Q.; Zhou, P.P.; Wang, X.; et al. Indole alkaloids from a soil-derived Clonostachys rosea. J. Nat. Prod. 2021, 84, 2468–2474.
- Knestrick, M.A.; Wilson, N.G.; Roth, A.; Adams, J.H.; Baker, B.J. Friomaramide, a highly modified linear hexapeptide from an Antarctic sponge, inhibits Plasmodium falciparum liver-stage development. J. Nat. Prod. 2019, 82, 2354–2358.
- Alcover, C.F.; Bernadat, G.; Kabran, F.A.; Le Pogam, P.; Leblanc, K.; Ramos, A.E.F.; Gallard, J.F.; Mouray, E.; Grellier, P.; Poupon, E.; et al. Molecular networking reveals serpentinine-related bisindole alkaloids from Picralima nitida, a previously well-investigated species. J. Nat. Prod. 2020, 83, 1207–1216.
- Otogo N’Nang, E.; Le Pogam, P.; Ndong Mba, T.; Sima Obiang, C.; Mouray, E.; Grellier, P.; Kumulungui, B.; Champy, P.; Beniddir, M.A. Targeted isolation of hemitheion from Mostuea brunonis, a proposed biosynthetic intermediate of theionbrunonines. J. Nat. Prod. 2021, 84, 1409–1413.
- Holland, D.C.; Prebble, D.W.; Er, S.; Hayton, J.B.; Robertson, L.P.; Avery, V.M.; Domanskyi, A.; Kiefel, M.J.; Hooper, J.N.A.; Carroll, A.R. α-Synuclein aggregation inhibitory prunolides and a dibrominated β-carboline sulfamate from the ascidian Synoicum prunum. J. Nat. Prod. 2022, 85, 441–452.
- Wang, Q.; Zhang, K.; Wang, W.; Zhang, G.; Zhu, T.; Che, Q.; Gu, Q.; Li, D. Amphiepicoccins A-J: Epipolythiodioxopiperazines from the fish-gill-derived Fungus Epicoccum nigrum HDN17-88. J. Nat. Prod. 2020, 83, 524–531.
- Li, J.; Hu, Y.; Hao, X.; Tan, J.; Li, F.; Qiao, X.; Chen, S.; Xiao, C.; Chen, M.; Peng, Z.; et al. Raistrickindole A, an anti-HCV oxazinoindole alkaloid from Penicillium raistrickii IMB17-034. J. Nat. Prod. 2019, 82, 1391–1395.
- Guo, Y.W.; Liu, X.J.; Yuan, J.; Li, H.J.; Mahmud, T.; Hong, M.J.; Yu, J.C.; Lan, W.J. L-Tryptophan induces a marine-derived Fusarium sp. to produce indole alkaloids with activity against the Zika virus. J. Nat. Prod. 2020, 83, 3372–3380.
- Zhang, Y.H.; Li, L.; Li, Y.Q.; Luo, J.H.; Li, W.; Li, L.F.; Zheng, C.J.; Cao, F. Oxalierpenes A and B, unusual indole-diterpenoid derivatives with antiviral activity from a marine-derived strain of the fungus Penicillium oxalicum. J. Nat. Prod. 2022, 85, 1880–1885.
- Zhou, L.M.; Kong, F.D.; Fan, P.; Ma, Q.Y.; Xie, Q.Y.; Li, J.H.; Zheng, H.Z.; Zheng, Z.H.; Yuan, J.Z.; Dai, H.F.; et al. Indole-diterpenoids with protein tyrosine phosphatase inhibitory activities from the marine-derived fungus Penicillium sp. KFD28. J. Nat. Prod. 2019, 82, 2638–2644.
- Di, X.; Wang, S.; Oskarsson, J.T.; Rouger, C.; Tasdemir, D.; Hardardottir, I.; Freysdottir, J.; Wang, X.; Molinski, T.F.; Omarsdottir, S. Bromotryptamine and imidazole alkaloids with anti-inflammatory activity from the Bryozoan Flustra foliacea. J. Nat. Prod. 2020, 83, 2854–2866.
- Shi, B.B.; Ai, H.L.; Duan, K.T.; Feng, T.; Liu, J.K. Ophiorrhines F and G, key biogenetic intermediates of ophiorrhine alkaloids from Ophiorrhiza japonica and their immunosuppressant activities. J. Nat. Prod. 2022, 85, 453–457.
- Jin, P.; Zhan, G.; Zheng, G.; Liu, J.; Peng, X.; Huang, L.; Gao, B.; Yuan, X.; Yao, G. Gelstriamine A a triamino monoterpene indole alkaloid with a caged 6/5/7/6/6/5 scaffold and analgesic alkaloids from Gelsemium elegans stems. J. Nat. Prod. 2021, 84, 1326–1334.
- Liang, J.H.; Luan, Z.L.; Tian, X.G.; Zhao, W.Y.; Wang, Y.L.; Sun, C.P.; Huo, X.K.; Deng, S.; Zhang, B.J.; Zhang, Z.J.; et al. Uncarialins A-I, monoterpenoid indole alkaloids from Uncaria rhynchophylla as natural agonists of the 5-HT1A receptor. J. Nat. Prod. 2019, 82, 3302–3310.
- Guo, Q.; Si, X.; Shi, Y.; Yang, H.; Liu, X.; Liang, H.; Tu, P.; Zhang, Q. Glucoconjugated monoterpene indole alkaloids from Uncaria rhynchophylla. J. Nat. Prod. 2019, 82, 3288–3301.
- Kong, F.D.; Zhang, S.L.; Zhou, S.Q.; Ma, Q.Y.; Xie, Q.Y.; Chen, J.P.; Li, J.H.; Zhou, L.M.; Yuan, J.Z.; Hu, Z.; et al. Quinazoline-containing indole alkaloids from the marine-derived fungus Aspergillus sp. HNMF114. J. Nat. Prod. 2019, 82, 3456–3463.
- Zhou, Y.F.; Hu, K.; Wang, F.; Tang, J.W.; Zhang, L.; Sun, H.D.; Cai, X.H.; Puno, P.T. 3-Hydroxy-4-methyldecanoic acid-containing cyclotetradepsipeptides from an endolichenic Beauveria sp. J. Nat. Prod. 2021, 84, 1244–1253.
- Li, C.J.; Chen, P.N.; Li, H.J.; Mahmud, T.; Wu, D.L.; Xu, J.; Lan, W.J. Potential antidiabetic fumiquinazoline alkaloids from the marine-derived fungus Scedosporium apiospermum F41-1. J. Nat. Prod. 2020, 83, 1082–1091.
- Klein-Junior, L.C.; Cretton, S.; Heyden, Y.V.; Gasper, A.L.; Nejad-Ebrahimi, S.; Christen, P.; Henriques, A.T. Bioactive azepine-indole alkaloids from Psychotria nemorosa. J. Nat. Prod. 2020, 83, 852–863.
- Zhai, Y.J.; Huo, G.M.; Zhang, Q.; Li, D.; Wang, D.C.; Qi, J.Z.; Han, W.B.; Gao, J.M. Phaeosphaones: Tyrosinase inhibitory thiodiketopiperazines from an endophytic Phaeosphaeria fuckelii. J. Nat. Prod. 2020, 83, 1592–1597.
- Perez-Bonilla, M.; Oves-Costales, D.; Gonzalez, I.; de la Cruz, M.; Martin, J.; Vicente, F.; Genilloud, O.; Reyes, F. Krisynomycins, imipenem potentiators against methicillin-resistant Staphylococcus aureus, Produced by Streptomyces canus. J. Nat. Prod. 2020, 83, 2597–2606.
This entry is offline, you can click here to edit this entry!