Toll-like receptor 4 is a protein that in humans is encoded by the TLR4 gene. TLR4 is a transmembrane protein, member of the toll-like receptor family, which belongs to the pattern recognition receptor (PRR) family. Its activation leads to an intracellular signaling pathway NF-κB and inflammatory cytokine production which is responsible for activating the innate immune system. TRL4 expressing cells are myeloid (erythrocytes, granulocytes, macrophages) rather than lymphoid (T-cells, B-cells, NK cells). Most myeloid cells also express high levels of CD14, which facilitates activation of TLR4 by LPS. It is most well known for recognizing lipopolysaccharide (LPS), a component present in many Gram-negative bacteria (e.g. Neisseria spp.) and selected Gram-positive bacteria. Its ligands also include several viral proteins, polysaccharide, and a variety of endogenous proteins such as low-density lipoprotein, beta-defensins, and heat shock protein. Palmitic acid is also a TLR4 agonist. TLR4 has also been designated as CD284 (cluster of differentiation 284). The molecular weight of TLR4 is approximately 95 kDa.
- lipopolysaccharide
- intracellular signaling
- toll-like receptor
1. Function
TLR4 is a member of the toll-like receptor (TLR) family, which plays a fundamental role in pathogen recognition and activation of innate immunity. They recognize pathogen-associated molecular patterns (PAMPs) that are expressed on infectious agents, and mediate the production of cytokines necessary for the development of effective immunity. TLRs are highly conserved from plants to Drosophila to humans and share structural and functional similarities.
The various TLRs exhibit different patterns of expression. This receptor is most abundantly expressed in placenta, and in myelomonocytic subpopulation of the leukocytes.
It cooperates with LY96 (also referred as MD-2) and CD14 to mediate in signal transduction events induced by lipopolysaccharide (LPS)[1] found in most gram-negative bacteria. Mutations in this gene have been associated with differences in LPS responsiveness.
TLR4 signaling responds to signals by forming a complex using an extracellular leucine-rich repeat domain (LRR) and an intracellular toll/interleukin-1 receptor (TIR) domain. LPS stimulation induces a series of interactions with several accessory proteins which form the TLR4 complex on the cell surface. LPS recognition is initiated by an LPS binding to an LBP protein. This LPS-LBP complex transfers the LPS to CD14. CD14 is a glycosylphosphatidylinositol-anchored membrane protein that binds the LPS-LBP complex and facilitates the transfer of LPS to MD-2 protein, which is associated with the extracellular domain of TLR4. LPS binding promotes the dimerization of TLR4/MD-2. The conformational changes of the TLR4 induce the recruitment of intracellular adaptor proteins containing the TIR domain which is necessary to activate the downstream signaling pathway.[2]
Several transcript variants of this gene have been found, but the protein-coding potential of most of them is uncertain.[3]
Most of the reported effects of TLR4 signaling in tumors are pro-carcinogenic mainly due to contributions of proinflammatory cytokine signaling (whose expression is driven by TLR-mediated signals) to tumor-promoting microenvironment.[4]
2. Signaling
Upon LPS recognition, conformational changes in the TLR4 receptors result in recruitment of intracellular TIR-domains containing adaptor molecules. These adaptors are associated with the TLR4 cluster via homophilic interactions between the TIR domains. There are four adaptor proteins involved in two major intracellular signaling pathways.[5]
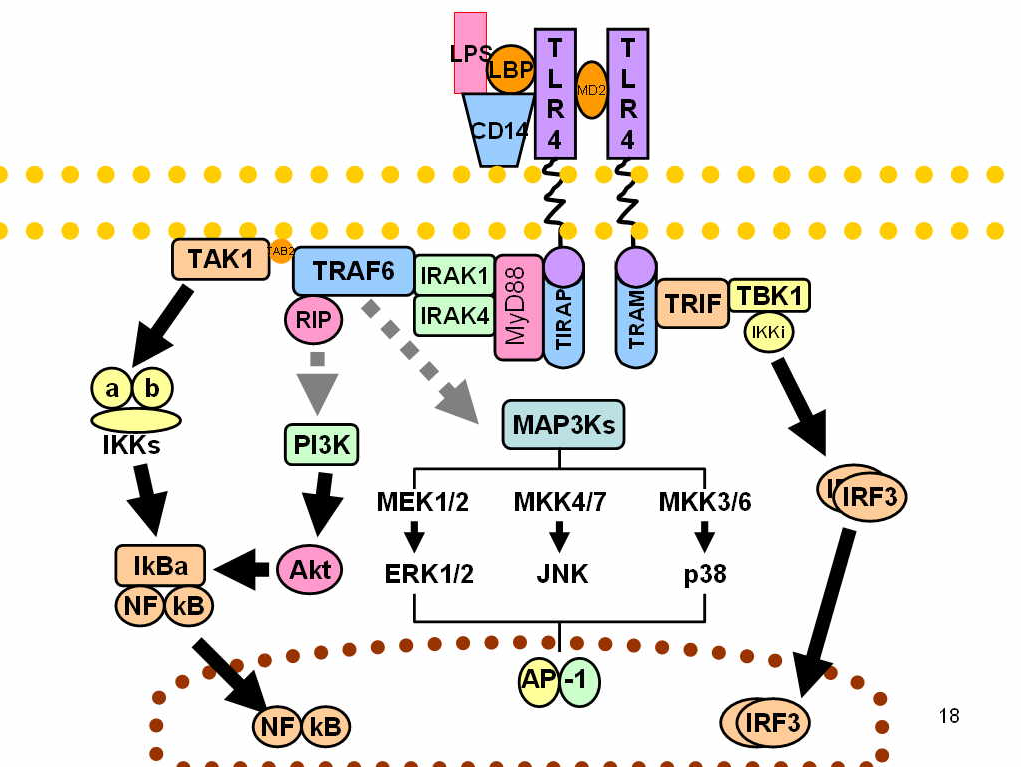
2.1. MyD88 – Dependent Pathway
The MyD88-dependent pathway is regulated by two adaptor-associated proteins: Myeloid Differentiation Primary Response Gene 88 (MyD88) and TIR Domain-Containing Adaptor Protein (TIRAP). TIRAP-MyD88 regulates early NF-κβ activation and production of proinflammatory cytokines, such as IL-12.[6] MyD88 signaling involves the activation of IL-1 Receptor-Associated Kinases (IRAKs) and the adaptor molecules TNF Receptor-Associated Factor 6 (TRAF6). TRAF6 induces the activation of TAK1 (Transforming growth factor-β-Activated Kinase 1) that leads to the activation of MAPK cascades (Mitogen-Activated Protein Kinase) and IKK (IκB Kinase). IKKs' signaling pathway leads to the induction of the transcription factor NF-κB, while activation of MAPK cascades lead to the activation of another transcription factor AP-1. Both of them have a role in the expression of proinflammatory cytokines.[2] The activation of NF-κB via TAK-1 is complex, and it starts by the assembly of a protein complex called the signalosome, which is made of a scaffolding protein, called NEMO. The protein complex is made from two different κB kinases, called IKKα and IKKβ. This causes the addition of a small regulatory protein to the signalosome called ubiquitin, that acts to initiate the release of the NF-κB protein, which coordinates translocation in the nucleus of cytokines.[7]
2.2. MyD88 – Independent Pathway
This TRIF-dependent pathway involves the recruitment of the adaptor proteins TIR-domain-containing adaptor inducing interferon-β (TRIF) and TRIF-related Adaptor Molecule (TRAM). TRAM-TRIF signals activate the transcription factor Interferon Regulatory Factor-3 (IRF3) via TRAF3. IRF3 activation induces the production of type 1 interferons.[5]
2.3. SARM – TRIF-Mediated Pathway
A fifth TIR-domain-containing adaptor protein called Sterile α and HEAT (Armadillo motif) (SARM) is a TLR4 signaling pathway inhibitor. SARM activation by LPS-binding inhibits -TRIF-mediated pathways but does not inhibit MyD88-mediated pathways. This mechanism prevents an excessive activation in response to LPS which may lead to inflammation-induced damage such as sepsis.[2]
3. Evolutionary History
TLR4 originated when TLR2 and TLR4 diverged about 500 million years ago near the beginning of vertebrate evolution.[8] Sequence alignments of human and great ape TLR4 exons have demonstrated that not much evolution has occurred in human TLR4 since our divergence from our last common ancestor with chimpanzees; human and chimp TLR4 exons only differ by three substitutions while humans and baboons are 93.5% similar in the extracellular domain.[9] Notably, humans possess a greater number of early stop codons in TLR4 than great apes; in a study of 158 humans worldwide, 0.6% had a nonsense mutation.[10][11] This suggests that there are weaker evolutionary pressures on the human TLR4 than on our primate relatives. The distribution of human TLR4 polymorphisms matches the out-of-Africa migration, and it is likely that the polymorphisms were generated in Africa before migration to other continents.[11][12]
4. Interactions
TLR4 has been shown to interact with:
Intracellular trafficking of TLR4 is dependent on the GTPase Rab-11a, and knock down of Rab-11a results in hampered TLR4 recruitment to E. coli-containing phagosomes and subsequent reduced signal transduction through the MyD88-independent pathway.[21]
5. Clinical Significance
Various single nucleotide polymorphisms (SNPs) of the TLR4 in humans have been identified[22] and for some of them an association with increased susceptibility to Gram-negative bacterial infections [23] or faster progression and a more severe course of sepsis in critically ill patients was reported.[24]
5.1. In Sepsis
TRL4 can be activated by binding to the lipid A portion of lipopolysaccharide found in Gram-negative bacteria.[25] Exaggerated and uncontrolled inflammation triggered by TRL4 during infection can lead to sepsis and septic shock.[26] Infections with Gram-negative bacteria such as Escherichia coli and Pseudomonas aeruginosa are the prevailing causes of severe sepsis in humans.[26]
5.2. In Insulin Resistance
Fetuin-A facilitates the binding of lipids to receptors, thereby contributing to insulin resistance.[27]
5.3. In Cancer Progression
TLR4 expression can be detected on many tumor cells and cell lines. TLR4 is capable of activating MAPK and NF-κB pathways, implicating possible direct role of cell-autonomous TLR4 signaling in regulation of carcinogenesis, in particular, through increased proliferation of tumor cells, apoptosis inhibition and metastasis. TLR4 signaling may also contribute to resistance to paclitaxel chemotherapy in ovary cancer and siRNA therapy in prostate cancer. 63% of breast cancer patients were reported to express TLR4 on tumor cells and the level of expression inversely correlated with the survival. Additionally, low MyD88 expression correlated with decreased metastasis to the lung and decreased CCL2 and CCL5 expression. TLR4 expression levels were the highest among TLRs in human breast cancer cell line MDA-MB-231 and TLR4 knockdown resulted in decreased proliferation and decreased IL-6 and IL-8 levels. On the other hand, TLR4 signaling in immune and inflammatory cells of tumor microenvironment may lead to production of proinflammatory cytokines (TNF, IL-1β, IL-6, IL-18, etc.), immunosuppressive cytokines (IL-10, TGF-β, etc.) and angiogenic mediators (VEGF, EGF, TGF-β, etc.).
These activities may result in further polarization of tumor-associated macrophages, conversion of fibroblasts into tumor-promoting cancer-associated fibroblasts, conversion of dendritic cells into tumor-associated DCs and activation of pro-tumorigenic functions of immature myeloid cells - Myeloid-derived Suppressor Cells (MDSC). TLR signaling has been linked to accumulation and function of MDSC at the site of tumor and it also allows mesenchymal stromal cells to counter NK cell-mediated anti-tumor immunity. In HepG2 hepatoblastoma cells LPS increased TLR4 levels, cell proliferation and resistance to chemotherapy, and these phenomena could be reversed by TLR4 gene knockdown. Similarly, LPS stimulation of human liver cancer cell line H7402 resulted in TLR4 upregulation, NF-κB activation, TNF, IL-6 and IL-8 production and increased proliferation that could be reversed by signal transducer and STAT3 inhibition. Besides the successful usage of Bacillus Calmette–Guérin in the therapy of bladder cancer there are reports on the treatment of oral squamous cell carcinoma, gastric cancer and cervical cancer with lyophilized streptococcal preparation OK-432 and utilization of TLR4/TLR2 ligands – derivatives of muramyl dipeptide.[4]
TLR4 stimulates B-cell responsiveness to Pokeweed mitogen for proliferation which can play a role in inhibiting tumor development.[28]
5.4. In Pregnancy
Activation of TLR4 in intrauterine infections leads to deregulation of prostaglandin synthesis, leading to uterine smooth muscle contraction.
5.5. Asp299Gly Polymorphism
Classically, TLR4 is said to be the receptor for LPS, however TLR4 has also been shown to be activated by other kinds of lipids. Plasmodium falciparum, a parasite known to cause the most common and serious form of malaria that is seen primarily in Africa, produces glycosylphosphatidylinositol, which can activate TLR4.[29] Two SNPs in TLR4 are co-expressed with high penetrance in African populations (i.e. TLR-4-Asp299Gly and TLR-4-Thr399Ile). These Polymorphisms are associated with an increase in TLR4-Mediated IL-10 production—an immunomodulator—and a decrease in proinflammatory cytokines.[30] The TLR-4-Asp299Gly point mutation is strongly correlated with an increased infection rate with Plasmodium falciparum. It appears that the mutation prevents TLR4 from acting as vigorously against, at least some plasmodial infections. The malaria infection rate and associated morbidity are higher in TLR-4-Asp299Gly group, but mortality appears to be decreased. This may indicate that at least part of the pathogenesis of malaria takes advantage of cytokine production. By reducing the cytokine production via the TLR4 mutation, the infection rate may increase, but the number of deaths due to the infection seem to decrease.[29]
In addition, TLR4-D299G has been associated with aggressive colorectal cancer in humans. It has been shown that human colon adenocarcinomas from patients with TLR4-D299G were more frequently of an advanced stage with metastasis than those with wild-type TLR4. The same study demonstrated functionally that intestinal epithelial cells (Caco-2) expressing TLR4-D299G underwent epithelial-mesenchymal transition and morphologic changes associated with tumor progression, whereas intestinal epithelial cells expressing wild-type TLR4 did not.[31]
6. Animal Studies
A link between the TLR4 receptor and binge drinking has been suggested. When genes responsible for the expression of TLR4 and GABA receptors are manipulated in rodents that had been bred and trained to drink excessively, the animals showed a "profound reduction" in drinking behaviours.[32] Additionally, it has been shown that ethanol, even in the absence of LPS, can activate TLR4 signaling pathways.[33]
High levels of TLR4 molecules and M2 tumor-associated macrophages are associated with increased susceptibility to cancer growth in mice deprived of sleep. Mice genetically modified so that they could not produce TLR4 molecules showed normal cancer growth.[34]
7. Drugs Targeting TLR4
Toll-like receptor 4 has been shown to be important for the long-term side-effects of opioid analgesic drugs. Various μ-opioid receptor ligands have been tested and found to also possess action as agonists or antagonists of TLR4, with opioid agonists such as (+)-morphine being TLR4 agonists, while opioid antagonists such as naloxone were found to be TLR4 antagonists. Activation of TLR4 leads to downstream release of inflammatory modulators including TNF-α and Interleukin-1, and constant low-level release of these modulators is thought to reduce the efficacy of opioid drug treatment with time, and be involved in both the development of tolerance to opioid analgesic drugs,[35][36] and in the emergence of side-effects such as hyperalgesia and allodynia that can become a problem following extended use of opioid drugs.[37][38] Drugs that block the action of TNF-α or IL-1β have been shown to increase the analgesic effects of opioids and reduce the development of tolerance and other side-effects,[39][40] and this has also been demonstrated with drugs that block TLR4 itself.
The response of TLR4 to opioid drugs has been found to be enantiomer-independent, so the "unnatural" enantiomers of opioid drugs such as morphine and naloxone, which lack affinity for opioid receptors, still produce the same activity at TLR4 as their "normal" enantiomers.[41][42] This means that the unnatural enantiomers of opioid antagonists, such as (+)-naloxone, can be used to block the TLR4 activity of opioid analgesic drugs, while leaving the μ-opioid receptor mediated analgesic activity unaffected.[42][43][44] This may also be the mechanism behind the beneficial effect of ultra-low dose naltrexone on opioid analgesia.[45]
Morphine causes inflammation by binding to the protein lymphocyte antigen 96, which, in turn, causes the protein to bind to Toll-like receptor 4 (TLR4).[46] The morphine-induced TLR4 activation attenuates pain suppression by opioids and enhances the development of opioid tolerance and addiction, drug abuse, and other negative side effects such as respiratory depression and hyperalgesia. Drug candidates that target TLR4 may improve opioid-based pain management therapies.[47]
7.1. Agonists
- Buprenorphine[48]
- Carbamazepine[49]
- Ethanol[50]
- Fentanyl[48]
- Levorphanol[48]
- Lipopolysaccharides (LPS)[51]
- Methadone[48]
- Morphine[48]
- Oxcarbazepine[49]
- Oxycodone[48]
- Pethidine[48]
- Glucuronoxylomannan from Cryptococcus[52][53]
- Morphine-3-glucuronide (inactive at opioid receptors, so selective for TLR4 activation)[38][48]
- Tapentadol (combined full μ-opioid receptor agonist and norepinephrine reuptake inhibitor)
- "Unnatural" isomers such as (+)-morphine activate TLR4 but lack opioid receptor activity,[41] although (+)-morphine also shows activity as a sigma receptor agonist.[54]
7.2. Antagonists
- The lipid A analog eritoran acts as a TLR4 antagonist. (As of December 2009), it was being developed as a drug against severe sepsis.[55] However, in 2013, a news story said the results against sepsis were somewhat disappointing and that it was better used to treat certain cases of severe influenza. Although it does not treat the virus itself, it could be used against the massive immune reaction called cytokine storm which often occurs later in the infection and is a major cause of mortality from severe influenza.[56]
- Amitriptyline[49]
- Cyclobenzaprine[49]
- Ketotifen[49]
- Imipramine[49]
- Mianserin[49]
- Ibudilast[57]
- Pinocembrin[58]
- Resatorvid[59]
- M62812
- Naloxone[48]
- (+)-Naloxone ("unnatural" isomer, lacks opioid receptor affinity so selective for TLR4 inhibition)[42]
- Naltrexone[48]
- (+)-Naltrexone[48]
- LPS-RS[48]
- Propentofylline
- Pentoxifylline[60] (and downregulate TLR4 expression[61])
- Tapentadol (mixed agonist/antagonist)
- TLR4-IN-C34[62]
- Palmitoylethanolamide[63]
The content is sourced from: https://handwiki.org/wiki/Biology:TLR4
References
- "O00206 (TLR4_HUMAN)". Uniprot. https://www.uniprot.org/uniprot/O00206.
- "LPS/TLR4 signal transduction pathway". Cytokine 42 (2): 145–51. May 2008. doi:10.1016/j.cyto.2008.01.006. PMID 18304834. https://dx.doi.org/10.1016%2Fj.cyto.2008.01.006
- "Entrez Gene: TLR4 toll-like receptor 4". https://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=7099.
- "TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis". Cytokine 89: 127–135. January 2017. doi:10.1016/j.cyto.2016.01.021. PMID 26854213. https://dx.doi.org/10.1016%2Fj.cyto.2016.01.021
- null
- "A comparative review of toll-like receptor 4 expression and functionality in different animal species". Frontiers in Immunology 5: 316. 2014. doi:10.3389/fimmu.2014.00316. PMID 25071777. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4090903
- "Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4". Immunology 113 (2): 153–62. 2004. doi:10.1111/j.1365-2567.2004.01976.x. PMID 15379975. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1782563
- "Evolution of the TIR, tolls and TLRs: functional inferences from computational biology". Current Topics in Microbiology and Immunology 270: 1–21. 2002. doi:10.1007/978-3-642-59430-4_1. ISBN 978-3-642-63975-3. PMID 12467241. https://dx.doi.org/10.1007%2F978-3-642-59430-4_1
- "Phylogenetic variation and polymorphism at the toll-like receptor 4 locus (TLR4)". Genome Biology 1 (1): RESEARCH002. 2000. doi:10.1186/gb-2000-1-1-research002. PMID 11104518. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=31919
- "Different selective pressures shape the evolution of Toll-like receptors in human and African great ape populations". Human Molecular Genetics 22 (23): 4829–40. December 2013. doi:10.1093/hmg/ddt335. PMID 23851028. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3820138
- "Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense". PLOS Genetics 5 (7): e1000562. July 2009. doi:10.1371/journal.pgen.1000562. PMID 19609346. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2702086
- "The evolutionary history of TLR4 polymorphisms in Europe". Journal of Innate Immunity 4 (2): 168–75. 2012. doi:10.1159/000329492. PMID 21968286. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6741577
- "Monomeric recombinant MD-2 binds toll-like receptor 4 tightly and confers lipopolysaccharide responsiveness". The Journal of Biological Chemistry 277 (26): 23427–32. June 2002. doi:10.1074/jbc.M202554200. PMID 11976338. https://dx.doi.org/10.1074%2Fjbc.M202554200
- "MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4". The Journal of Experimental Medicine 189 (11): 1777–82. June 1999. doi:10.1084/jem.189.11.1777. PMID 10359581. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2193086
- "Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors". Nature Immunology 5 (5): 495–502. May 2004. doi:10.1038/ni1066. PMID 15107846. https://dx.doi.org/10.1038%2Fni1066
- "Toll-like receptor 3 mediates a more potent antiviral response than Toll-like receptor 4". Journal of Immunology 170 (7): 3565–71. April 2003. doi:10.4049/jimmunol.170.7.3565. PMID 12646618. https://dx.doi.org/10.4049%2Fjimmunol.170.7.3565
- "Murine TOLL-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NF kappa B and expression of the inducible cyclooxygenase". The Journal of Biological Chemistry 275 (44): 34035–40. November 2000. doi:10.1074/jbc.M007386200. PMID 10952994. https://dx.doi.org/10.1074%2Fjbc.M007386200
- "Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction". Nature 413 (6851): 78–83. September 2001. doi:10.1038/35092578. PMID 11544529. Bibcode: 2001Natur.413...78F. https://dx.doi.org/10.1038%2F35092578
- "Negative regulation of toll-like receptor-mediated signaling by Tollip". The Journal of Biological Chemistry 277 (9): 7059–65. March 2002. doi:10.1074/jbc.M109537200. PMID 11751856. https://dx.doi.org/10.1074%2Fjbc.M109537200
- "Ni(II) interaction with a peptide model of the human TLR4 ectodomain". J Trace Elem Med Biol 44: 151–160. 2017. doi:10.1016/j.jtemb.2017.07.006. PMID 28965571. https://dx.doi.org/10.1016%2Fj.jtemb.2017.07.006
- "The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes". Immunity 33 (4): 583–96. October 2010. doi:10.1016/j.immuni.2010.09.010. PMID 20933442. https://dx.doi.org/10.1016%2Fj.immuni.2010.09.010
- "Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious disease". The Lancet. Infectious Diseases 5 (3): 156–64. March 2005. doi:10.1016/S1473-3099(05)01308-3. PMID 15766650. https://dx.doi.org/10.1016%2FS1473-3099%2805%2901308-3
- "Relevance of mutations in the TLR4 receptor in patients with gram-negative septic shock". Archives of Internal Medicine 162 (9): 1028–32. May 2002. doi:10.1001/archinte.162.9.1028. PMID 11996613. https://dx.doi.org/10.1001%2Farchinte.162.9.1028
- "Polymorphisms of the toll-like receptor 2 and 4 genes are associated with faster progression and a more severe course of sepsis in critically ill patients". The Journal of International Medical Research 42 (1): 93–110. February 2014. doi:10.1177/0300060513504358. PMID 24366499. https://dx.doi.org/10.1177%2F0300060513504358
- "O-antigen structural variation: mechanisms and possible roles in animal/plant-microbe interactions". FEMS Microbiology Reviews 26 (1): 17–47. 2002. doi:10.1111/j.1574-6976.2002.tb00597.x. PMID 12007641. https://dx.doi.org/10.1111%2Fj.1574-6976.2002.tb00597.x
- "TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling". Cellular and Molecular Life Sciences 78 (4): 1233–1261. 2021. doi:10.1007/s00018-020-03656-y. ISSN 1420-682X. PMID 33057840. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=7904555
- "Effects of fetuin-A with diverse functions and multiple mechanisms on human health". Clinical Biochemistry 88: 1–10. February 2021. doi:10.1016/j.clinbiochem.2020.11.004. PMID 33245873. https://dx.doi.org/10.1016%2Fj.clinbiochem.2020.11.004
- "Poke weed mitogen requires Toll-like receptor ligands for proliferative activity in human and murine B lymphocytes". PLOS ONE 7 (1): e29806. 2012-01-04. doi:10.1371/journal.pone.0029806. PMID 22238657. Bibcode: 2012PLoSO...729806B. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3251602
- "Toll-like receptor (TLR) polymorphisms in African children: Common TLR-4 variants predispose to severe malaria". Proceedings of the National Academy of Sciences of the United States of America 103 (1): 177–82. January 2006. doi:10.1073/pnas.0506803102. PMID 16371473. Bibcode: 2006PNAS..103..177M. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1324982
- "Toll-like receptor 4 Asp299Gly/Thr399Ile polymorphisms are a risk factor for Candida bloodstream infection". European Cytokine Network 17 (1): 29–34. March 2006. PMID 16613760. http://www.jle.com/fr/revues/ecn/e-docs/toll_like_receptor_4_asp299gly_thr399ile_polymorphisms_are_a_risk_factor_for_candida_bloodstream_infection_268300/article.phtml.
- "Toll-like receptor 4 variant D299G induces features of neoplastic progression in Caco-2 intestinal cells and is associated with advanced human colon cancer". Gastroenterology 141 (6): 2154–65. December 2011. doi:10.1053/j.gastro.2011.08.043. PMID 21920464. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3268964
- "Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala". Proceedings of the National Academy of Sciences of the United States of America 108 (11): 4465–70. March 2011. doi:10.1073/pnas.1019020108. PMID 21368176. Bibcode: 2011PNAS..108.4465L. Lay summary in: "Genes associated with binge drinking identified". sciencedaily.com (Press release). March 1, 2011. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3060224
- "Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes". Journal of Immunology 175 (10): 6893–9. November 2005. doi:10.4049/jimmunol.175.10.6893. PMID 16272348. https://dx.doi.org/10.4049%2Fjimmunol.175.10.6893
- "Fragmented sleep accelerates tumor growth and progression through recruitment of tumor-associated macrophages and TLR4 signaling". Cancer Research 74 (5): 1329–37. March 2014. doi:10.1158/0008-5472.CAN-13-3014. PMID 24448240. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4247537
- "Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance". Pain 115 (1–2): 50–9. May 2005. doi:10.1016/j.pain.2005.02.003. PMID 15836969. https://dx.doi.org/10.1016%2Fj.pain.2005.02.003
- "Dual regulation of mu opioid receptors in SK-N-SH neuroblastoma cells by morphine and interleukin-1β: evidence for opioid-immune crosstalk". Journal of Neuroimmunology 227 (1–2): 26–34. October 2010. doi:10.1016/j.jneuroim.2010.06.007. PMID 20615556. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2942958
- Mechanism of allodynia evoked by intrathecal morphine-3-glucuronide in mice. International Review of Neurobiology. 85. 2009. pp. 207–19. doi:10.1016/S0074-7742(09)85016-2. ISBN 9780123748935. https://dx.doi.org/10.1016%2FS0074-7742%2809%2985016-2
- "Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta". Neuroscience 165 (2): 569–83. January 2010. doi:10.1016/j.neuroscience.2009.10.011. PMID 19833175. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2795035
- "Etanercept restores the antinociceptive effect of morphine and suppresses spinal neuroinflammation in morphine-tolerant rats". Anesthesia and Analgesia 112 (2): 454–9. February 2011. doi:10.1213/ANE.0b013e3182025b15. PMID 21081778. https://dx.doi.org/10.1213%2FANE.0b013e3182025b15
- "An IL-1 receptor antagonist blocks a morphine-induced attenuation of locomotor recovery after spinal cord injury". Brain, Behavior, and Immunity 25 (2): 349–59. February 2011. doi:10.1016/j.bbi.2010.10.018. PMID 20974246. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3025088
- "The "toll" of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia". Trends in Pharmacological Sciences 30 (11): 581–91. November 2009. doi:10.1016/j.tips.2009.08.002. PMID 19762094. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2783351
- "Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4)". The European Journal of Neuroscience 28 (1): 20–9. July 2008. doi:10.1111/j.1460-9568.2008.06321.x. PMID 18662331. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2588470
- "Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia". Brain, Behavior, and Immunity 22 (8): 1178–89. November 2008. doi:10.1016/j.bbi.2008.05.004. PMID 18599265. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2783238
- "Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences". Neuroscience 167 (3): 880–93. May 2010. doi:10.1016/j.neuroscience.2010.02.011. PMID 20178837. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2854318
- "Ultra-low dose naloxone upregulates interleukin-10 expression and suppresses neuroinflammation in morphine-tolerant rat spinal cords". Behavioural Brain Research 207 (1): 30–6. February 2010. doi:10.1016/j.bbr.2009.09.034. PMID 19799935. https://dx.doi.org/10.1016%2Fj.bbr.2009.09.034
- "Neuroscience: Making morphine work better". Nature 484 (7395): 419. 26 April 2012. doi:10.1038/484419a. Bibcode: 2012Natur.484Q.419.. https://dx.doi.org/10.1038%2F484419a
- Drahl C (22 August 2012). "Small Molecules Target Toll-Like Receptors". Chemical & Engineering News. http://cen.acs.org/articles/90/web/2012/08/Small-Molecules-Target-Toll-Like.html.
- "Evidence that opioids may have toll-like receptor 4 and MD-2 effects". Brain, Behavior, and Immunity 24 (1): 83–95. January 2010. doi:10.1016/j.bbi.2009.08.004. PMID 19679181. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2788078
- "Evidence that tricyclic small molecules may possess toll-like receptor and myeloid differentiation protein 2 activity". Neuroscience 168 (2): 551–63. June 2010. doi:10.1016/j.neuroscience.2010.03.067. PMID 20381591. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2872682
- "Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage". Brain, Behavior, and Immunity 25 (Suppl 1): S80–91. June 2011. doi:10.1016/j.bbi.2011.02.012. PMID 21352907. https://dx.doi.org/10.1016%2Fj.bbi.2011.02.012
- "Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders". Brain, Behavior, and Immunity 25 (Suppl 1): S13–20. June 2011. doi:10.1016/j.bbi.2010.12.013. PMID 21193024. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4068736
- "Percutaneous penetration of 2,4-dichlorophenoxyacetic acid and 2,4-D dimethylamine salt in human volunteers". Journal of Toxicology and Environmental Health 36 (3): 233–40. July 1992. doi:10.1080/15287399209531634. PMID 1629934. https://dx.doi.org/10.1080%2F15287399209531634
- "Glucuronoxylomannan, a microbial compound, regulates expression of costimulatory molecules and production of cytokines in macrophages". The Journal of Infectious Diseases 191 (1): 127–37. January 2005. doi:10.1086/426511. PMID 15593014. https://dx.doi.org/10.1086%2F426511
- "Stereoselective action of (+)-morphine over (-)-morphine in attenuating the (-)-morphine-produced antinociception via the naloxone-sensitive sigma receptor in the mouse". European Journal of Pharmacology 571 (2–3): 145–51. October 2007. doi:10.1016/j.ejphar.2007.06.012. PMID 17617400. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2080825
- "Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis". Critical Care Medicine 38 (1): 72–83. January 2010. doi:10.1097/CCM.0b013e3181b07b78. PMID 19661804. https://dx.doi.org/10.1097%2FCCM.0b013e3181b07b78
- "New drug offers novel approach to taming flu virus - Vitals". NBCNews. 1 May 2013. http://vitals.nbcnews.com/_news/2013/05/01/18002315-new-drug-offers-novel-approach-to-taming-flu-virus.
- "[Toll-like receptor 4: the potential therapeutic target for neuropathic pain]". Zhongguo Yi Xue Ke Xue Yuan Xue Bao. Acta Academiae Medicinae Sinicae 34 (2): 168–73. April 2012. doi:10.3881/j.issn.1000-503X.2012.02.013. PMID 22776604. https://dx.doi.org/10.3881%2Fj.issn.1000-503X.2012.02.013
- "Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia". Brain, Behavior, and Immunity 61: 326–339. March 2017. doi:10.1016/j.bbi.2016.12.012. PMID 28007523. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5453178
- "Structure-Based Design, Synthesis, and Biological Evaluation of Imidazo[4,5-b]Pyridin-2-one-Based p38 MAP Kinase Inhibitors: Part 2". ChemMedChem 14 (24): 2093–2101. December 2019. doi:10.1002/cmdc.201900373. PMID 31697454. https://dx.doi.org/10.1002%2Fcmdc.201900373
- "Pentoxifylline inhibits TLR- and inflammasome-mediated in vitro inflammatory cytokine production in human blood with greater efficacy and potency in newborns". Pediatric Research 81 (5): 806–816. May 2017. doi:10.1038/pr.2017.6. PMID 28072760. https://dx.doi.org/10.1038%2Fpr.2017.6
- "Pentoxifylline modulates LPS-induced hyperinflammation in monocytes of preterm infants in vitro". Pediatric Research 82 (2): 215–225. August 2017. doi:10.1038/pr.2017.41. PMID 28288151. https://dx.doi.org/10.1038%2Fpr.2017.41
- Neal MD, Jia H, Eyer B, Good M, Guerriero CJ, Sodhi CP, Afrazi A, Prindle T Jr, Ma C, Branca M, Ozolek J, Brodsky JL, Wipf P, Hackam DJ. Discovery and validation of a new class of small molecule Toll-like receptor 4 (TLR4) inhibitors. PLOS ONE. 2013 Jun 12;8(6):e65779. doi:10.1371/journal.pone.0065779 PMID 23776545 https://doi.org/10.1371%2Fjournal.pone.0065779
- "Ultramicronized palmitoylethanolamide reduces inflammation an a Th1-mediated model of colitis". European Journal of Inflammation 13: 14–31. 2015. doi:10.1177/1721727X15575869. https://dx.doi.org/10.1177%2F1721727X15575869