Glaucoma is a progressive age-related disease of the visual system and the leading cause of irreversible blindness worldwide. Currently, intraocular pressure (IOP) is the only modifiable risk factor for the disease, but even as IOP is lowered, the pathology of the disease often progresses. Hence, effective clinical targets for the treatment of glaucoma remain elusive. Glaucoma shares comorbidities with a multitude of vascular diseases, and evidence in humans and animal models demonstrates an association between vascular dysfunction of the retina and glaucoma pathology. Integral to the survival of retinal ganglion cells (RGCs) is functional neurovascular coupling (NVC), providing RGCs with metabolic support in response to neuronal activity. NVC is mediated by cells of the neurovascular unit (NVU), which include vascular cells, glial cells, and neurons. Nitric oxide-cyclic guanosine monophosphate (NO-cGMP) signaling is a prime mediator of NVC between endothelial cells and neurons, but emerging evidence suggests that cGMP signaling is also important in the physiology of other cells of the NVU. NO-cGMP signaling has been implicated in glaucomatous neurodegeneration in humans and mice.
1. Introduction
Glaucoma is the leading cause of irreversible blindness worldwide, with global prevalence of the disease estimated to surpass 100 million cases by 2040 [
1,
2]. Glaucoma describes an umbrella of optic neuropathies that lead to the progressive degeneration of retinal ganglion cells (RGCs) and their axons that form the optic projection [
3]. The disease is characterized by the increased sensitivity of RGCs and their axons to intraocular pressure (IOP), with degeneration of RGCs occurring sectorially over time; i.e., from one retinotopic sector to the next [
4,
5]. Current therapies aim to lower IOP using topical medication or surgical intervention; however, this is not effective for all patients and many progress to permanent vision loss. Moreover, a proportion of patients are normotensive, with IOPs within the typical range, and RGC axon degeneration still persists [
6].
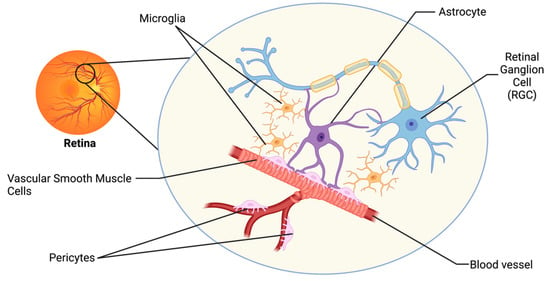
2. The Retinal Neurovascular Unit and Glaucoma
The visual system has developed intricate regulatory mechanisms to fulfill the high metabolic demand of the retina. One such mechanism is neurovascular coupling (NVC), whereby an increased neuronal activity promotes signaling between cells of the neurovascular unit (NVU), resulting in increased supply of metabolites and removal of waste products by the vasculature [
12]. The retinal NVU consists of three main cell types: neurons, glial cells (including astrocytes, microglia, and oligodendrocytes), and vascular cells (endothelial cells, vascular smooth muscle cells, and pericytes;
Figure 1) [
13]. Dysfunction in the NVU is evident in the pathophysiology of many neurodegenerative diseases of the central nervous system (CNS), including Alzheimer’s Disease and Parkinson’s Disease [
3]. Such neurodegenerative diseases share many common pathophysiologies with glaucoma, and thus dysfunction in the NVU may also be a relevant mechanism of neurodegeneration of the visual system [
3,
13].
Figure 1. The retinal neurovascular unit. The retinal NVU consists of three main cell types: neurons (retinal ganglion cells; RGCs), glial cells (including astrocytes and microglia), and vascular cells (including endothelial cells, vascular smooth muscle cells, and pericytes).
Vascular autoregulation and NVC mechanisms are severely compromised in glaucoma patients [
14]. Dysfunction of the NVC response to light flicker was perturbed in patients with early-stage glaucoma progression, with only moderate IOP increases [
14]. The direct effect of IOP on light-flicker-induced NVC was further tested using patients exposed to short-term induced IOP [
15]; however, no changes in NVC response were observed. The change in NVC evident in early-stage glaucoma patients could be due to reduced function of RGCs or due to the dysfunction of cells in the NVU, which remains undetectable in the clinic. Rodent models of glaucoma have been a valuable resource in trying to better understand neurovascular mechanisms of the disease.
3. NO-cGMP Signaling in Glaucoma
The nitric oxide-cyclic guanosine monophosphate (NO-cGMP) signaling pathway is integral to NVC mechanisms and is the prime mediator of vasodilation in endothelial cells [
9]. NO is produced by nitric oxide synthase enzymes in cells of the NVU in both physiological and pathophysiological conditions [
17]. NO is lipophilic and can freely traverse cellular membranes, where it binds to its primary receptor, soluble guanylate cyclase (GC1; [
18,
19]). GC1 binding of NO stimulates the production of cGMP, a second messenger capable of binding to multiple cellular protein targets such as cyclic nucleotide phosphodiesterases (PDEs), cGMP-dependent protein kinases, and cGMP-gated ion channels [
20,
21,
22,
23]. cGMP signaling is multifaceted, with the potential to exert broad effects on cellular physiology, depending upon the cellular target that it binds.
Dysfunction in cGMP signaling has been implicated in glaucoma pathophysiology in human patients and in animal models of the disease. Levels of NO and cGMP in aqueous humor and plasma are reduced in glaucoma patients [
24,
25]. Furthermore, in a genome-wide association study (GWAS) of patients with primary open angle glaucoma (POAG), a single nucleotide polymorphism (rs11722059) located in the
GUCY1A3/GUCY1B3 gene locus was significantly associated with paracentral visual field loss in female glaucoma patients [
26].
GUCY1A3/GUCY1B3 encode the alpha and beta subunits of GC1, respectively [
26]. Identification of a risk variant in the GC1 gene and expression of GC1 in human and rodent ocular tissues including RGCs [
26] suggests that GC1 may have a role in the pathogenesis of glaucoma.
4. NO-cGMP Signaling in Vascular Cells
The vascular cells of the NVU in the brain consist of the endothelial cells, vascular smooth muscle cells, and pericytes. Nestled between the blood vessel lumen and the smooth muscle cells, the endothelium is responsible for modulation of vascular tone, thrombus formation, cell adhesion, smooth muscle proliferation, and sequestration of inflammatory factors [
33]. Within the retinal NVU, the function is similar; endothelial cells line retinal vessels responsible for RGC blood supply and smooth muscle cells control vascular tone [
9,
34]. Critical to endothelial function is cGMP signaling; outside of the CNS, cGMP signaling plays a major role in decreasing endothelial cell permeability and is responsible for smooth muscle cell relaxation and subsequent vasodilation [
35,
36].
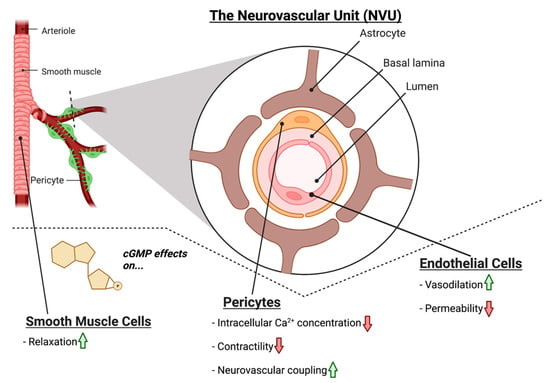
Within the NVU, NO-cGMP signaling plays a similar role in modulating vascular tone; NO produced in the endothelium acts on GC1 in smooth muscle cells to induce vasodilation via smooth muscle relaxation (
Figure 2) [
34]. Moreover, cGMP is also implicated in decreasing the permeability of the endothelium of the NVU. Experimental treatment with the cGMP agonist 8-Br-cGMP decreased the permeability of an in vitro blood–brain barrier (BBB) model [
37].
Figure 2. The effects of cGMP on vascular cells of the retinal NVU. cGMP increases smooth muscle cell relaxation. In pericytes, cGMP has been shown to reduce intracellular Ca2+, decrease contractility, and increase NVC mechanisms. In endothelial cells, cGMP has been shown to increase vasodilation, while reducing endothelial cell permeability.
The vascular cells of the NVU also include pericytes; however, exploration of their role within the retinal NVU in glaucoma pathology is in its infancy compared to other cells of the retinal vasculature [
9]. Pericytes are situated in the basement membrane of microvessels and are responsible for a multitude of functions, including maintaining vascular stability, providing structural support for the blood vessels, mediating vasodilation and vasoconstriction, and BBB maintenance [
13,
45,
46,
47,
48,
49]. The tone of pericyte-containing microvessels is largely controlled by the balance between Ca
2+-mediated contractility and NO-mediated relaxation [
50]. Recently, a key role for pericytes in retinal NVC and glaucoma pathology was uncovered [
16]. The identification of inter-pericyte tunneling nanotubes (IP-TNTs) between pericyte cells in the retina led to the discovery that pericytes use IP-TNTs to coordinate vascular responses to neuronal activity across the retina [
51]. Damage to IP-TNTs in glaucoma impairs light-evoked neurovascular coupling via persistent Ca
2+ influx, causing vasoconstriction [
16].
The role of cGMP signaling in pericytes located outside of the CNS has been explored extensively and may provide a mechanistic link between retinal pericyte dysfunction (specifically pericyte-associated vasoconstriction and NVC breakdown) and the vascular-associated glaucomatous pathology observed in patients. In liver pericytes, exogenous NO supplementation and inhibition of cGMP breakdown via PDE inhibitor sildenafil citrate diminished the extent of endothelial dysfunction caused by contraction of active pericytes [
56].
5. NO-cGMP Signaling in Glial Cells
The retinal glia, including astrocytes, Müller glia, and microglia, were thought to be relatively passive elements in the CNS; however, recently, it has become clear that they play a critical intermediary role between the neurons and vascular cells of the NVU [
13,
61,
62]. The predominant glial cells in the retina are the Müller glia (MG), which span the retinal layers, from the inner border with the vitreous humor, to the photoreceptor layer. Astrocytes are present in the superficial layer of the retina and make intimate connections between RGCs and blood vessels. Astrocytes and MG together form the blood–retinal barrier (BRB) and are integral to retinal NVC [
13,
63,
64]. Astrocytes play a critical role in shuttling energy resources to neurons, responding to glutamatergic activity by producing lactate and releasing it into the extracellular space, which neurons then utilize for metabolic demands [
65,
66,
67]. Astrocytes also regulate neurotransmitter release and re-uptake, to help maintain neuronal homeostasis [
13,
68,
69,
70,
71]. In humans, regarding glial cells, the fovea contains only MG subtypes, indicating that these cells alone are sufficient to supply the metabolic needs of the RGCs [
13,
71]. Microglia are also resident glial cells that span multiple layers of the retina, yet their exact role in the basal NVU function of the retina is less understood compared to other glial cell types [
72].
In glaucoma, glial dysfunction and/or activation may contribute to breakdown in NVC, rendering RGCs more susceptible to degeneration. Given their localization in the RGC layer and role in metabolic support, changes in astrocyte physiology may have a large impact on the health and physiology of RGCs. During pathological conditions in the retina, astrocytes respond robustly with gross morphological and physiological changes, which may prompt the dysfunction and degeneration of RGC axons [
9,
67,
74,
75].
Examining the role of NO-cGMP signaling in the NVU may help to elucidate the relationship between glia, vascular function, and glaucoma pathology. In the CNS, decreased GC1 expression is detected in the reactive glia of post-mortem patients with neurodegenerative diseases such Alzheimer’s disease and multiple sclerosis [
84,
85,
86]. In cultured retinal MG, addition of cGMP analogues activates a calcium permeable, non-selective cation current via the opening of cGMP-gated channels, indicating that cGMP signaling, in part, regulates MG physiology [
87]. In the microglia, cGMP signaling pathways are involved in inflammatory gene expression [
84,
88,
89,
90,
91,
92,
93,
94]. Pharmacological inhibition of GC1 prevents activation and migration of microglia to injured or inflamed sites in the CNS; recruitment can be rescued with the addition of a cGMP analog, suggesting that microglial recruitment is, at least in part, mediated by cGMP signaling [
95,
96].
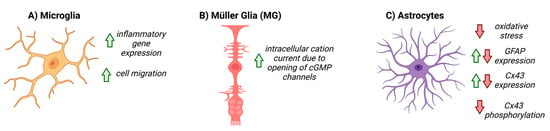
To date, the role of NO-cGMP signaling in astrocyte function has been explored in many contexts, both within the CNS and systemically, and has been summarized in
Figure 3. In cultured rat cerebellar astrocytes, cGMP directly affects the motility and morphology of cells and expression of glial fibrillary acidic protein (GFAP), suggesting that cGMP signaling directly modulates astrocyte physiology [
97,
98]. In focal brain injury, inhibiting PDE increased cGMP and altered the glial inflammatory response, decreased oxidative stress and cell death, and increased angiogenesis (
Figure 3) [
84].
Figure 3. The known effects of cGMP on glial cells. Red arrows represent downregulation, or negative activity, green arrows represent increased expression, or positive activity. (A) cGMP upregulates inflammatory gene expression and cell migration in microglia, (B) increases cation current within Müller cells via opening of cGMP-gated channels, and (C) decreases oxidative stress, differentially affects GFAP and Cx43 expression, and decreases Cx43 phosphorylation in astrocytes.
Moreover, changes in astrocyte connexin expression have also been observed in
GC1−/− mice [
29]. Connexin-43 (Cx43) is the most prevalent connexin in astrocytes, and
GC1−/− mice exhibit a greater decrease in Cx43 levels with age [
29,
67]. Connexins are crucial in astrocyte cell communication in the NVU, and increases in pressure in vitro cause a decrease in gap-junction communication via alterations in connexin activity [
77]. cGMP/PKG activity has been shown to alter connexin activity, specifically through phosphorylation, hinting at a link between cGMP activity, connexins, and astrocyte function in the NVU [
100]. cGMP-gated channels also impart changes in astrocyte physiology that may perturb vascular function in glaucoma. cGMP-gated Ca
2+ channels have been confirmed to populate the astrocytes of the rat forebrain, midbrain, and hindbrain, and are important for astrocyte function [
104]. MG also likely possess cGMP-gated Ca
2+ channels [
87]. Within the endfeet of astrocytes in the NVU, Ca
2+ signaling plays a critical role in maintaining the integrity of the BBB [
105]; yet, it is unknown if the cGMP-gated Ca
2+ channels specifically contribute to the abnormal vascular function of astrocytes in the glaucomatous NVU. cGMP-gated Ca
2+ channels in astrocytes have been investigated as a potential therapeutic target for vascular dysfunction in Alzheimer’s disease [
106]. Given that glaucoma shares many pathological commonalities with Alzheimer’s disease [
3], investigating the putative role of cGMP-gated Ca
2+ channels in retinal astrocytes in glaucoma may uncover a novel therapeutic target.
6. NO-cGMP Signaling in Retinal Neurons
In glaucoma, the degeneration of RGCs and their axons in the optic projection results in irreversible vision loss. The role of NO-cGMP signaling in RGCs appears to be multifaceted. In CNS neurons, NO-cGMP signaling is important in NVC, synaptic plasticity, and synaptic transmission [107]. GC1 is expressed in pre- and post-synaptic terminals and in neuronal soma and spines [107,108]. GC1 is also evident in RGC soma in the retina [26]. The depolarization of neuronal membranes stimulates Ca2+ influx into RGCs, which activates neuronal nitric oxide synthase, leading to NO production and release, stimulating vasodilation in its target vascular cells via cGMP [62]. Interestingly, GC1−/− mice [26] develop RGC degeneration with age, and increasing availability of cGMP has a neuroprotective effect in murine glaucoma models [28]. Furthermore, primary RGC cultures and mouse retinal explants exhibit decreased apoptotic signaling when supplemented with cGMP [109]. The transcription factor CREB is responsible for neuroprotective functions and is phosphorylated and activated by PKG in CNS neurons [110,119,120,121]. Alternatively, cGMP, via PKG, can also modulate intracellular calcium signaling in RGCs [122] and therefore may be responsible for limiting the neurotoxic effects to neurons of high intracellular Ca2+ levels. Since cGMP has multiple downstream effectors that may contribute to the neuroprotective effects observed in RGCs, further research is needed to distinguish the apparent neuroprotective and neurodegenerative arms of the cGMP signaling cascade.
This entry is adapted from the peer-reviewed paper 10.3390/biom12111671