2. Neurokinin-1 Receptor and Cancer
2.1. Neurokinin-1 Receptor: General Findings
NK-1R, also known as SP receptor or tachykinin 1 receptor, belongs to the rhodopsin-like G protein-coupled receptors family (seven-transmembrane domain receptors, serpentine receptors, or 7TM receptors). It is widely distributed by the whole body (e.g., nervous system, immune and endothelial cells, lung, gastrointestinal tract, skin), and it is highly conserved along the species [
17,
18]. The tachykinin receptor 1 (
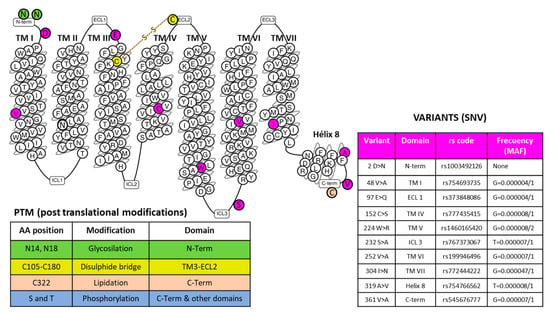
TACR1) gene with cytogenetic location 2p12 encodes the human NK-1R that expands through the plasma membrane, forming seven transmembrane domains and several intracellular and extracellular loops. The receptor’s function may be altered by anomalous expression, heterodimerization with other receptors, point mutations, or derangement of post-translational modifications. Specific amino acid positions may suffer post-translational changes, including glycosylation, formation of a disulfide bridge, and lipidation modification (
Figure 1). Hundreds of single nucleotide variants (SNVs) have been described [
19], but their pathological significance has not been analyzed thoroughly. They ask for further analysis related to specific pathologies, including cancer. Different classes of G protein-coupled receptors may assemble in operative homodimers, heterodimers, and oligomers [
20]. These macromolecular structures amplify and diversify signaling responses and, in some cases, may serve as specific therapeutic targets for several pathological entities [
20,
21]. No information on the physiological formation of NK-1R dimers or oligomers has been provided. NK-1R functions through functional monomers residing in membrane microdomains where cholesterol is essential [
22]. However, the capacity to heterodimerize appears possible. Artificially built heterodimers of NK-1R and β
2-adrenergic receptors were functional and internalized as a complex [
23]. Therefore, the formation of complexes with the intervention of NK-1R may have relevance for the homeostasis of NK-1R signaling in abnormal or adaptive circumstances and requires further exploration.
Figure 1. The snake plot of the human NK-1R (modified from [
24]) highlights amino acid positions involved in post-translational modifications (PTM) and representative SNVs (Single Nucleotide Variations) in different receptor domains, as indicated. The table on the right-hand side of the figure showing the example variants depicts the amino acid position and change, the domain localization, the registered rs code, and the frequency acquired from the Single Nucleotide Polymorphism Database (dSNP) [
19]. Abbreviations: ECL, extracellular loops; ICL, intracellular loops; MAF, minor allele frequency; TM, a transmembrane domain.
Seven-transmembrane helix receptors show a carboxy-terminal cytoplasmic domain (involved in the desensitization of the receptor), an amino-terminal extracellular domain (involved in the specificity of the receptor), and three intracellular and extracellular loops flanked by seven intermembrane domains [
25]. The extracellular N-terminus and the intracellular C-terminus contain asparagine glycosylation and serine/threonine phosphorylation sites, respectively, which regulate NK-1R signaling [
25]. NK-1R shows different active conformations, each of which has a different affinity for distinct agonists or antagonists: SP binds to the extracellular loops, and non-peptide NK-1R antagonists bind deep between the III and VI transmembrane segments of NK-1R [
25]. Specific residues of this receptor (e.g., Gln165, His197, and 265) regulate the binding of non-peptide NK-1R antagonists [
25]. NK-1R can be coupled to Gαq, Gαs, Gαi, Gαo, and Gα
12/13 proteins, the activation of a determined G protein being regulated by the type of ligands and NK-1R conformation [
25,
26,
27,
28,
29]. G proteins differ in the effectors/signaling pathways they activate, and via these pathways, the transcription of specific genes is controlled [
2]. Gαi blocks adenylate cyclase activity, decreases the level of cyclic adenosine monophosphate, and increases the phosphorylation of extracellular signal-regulated kinases. Gαs activates adenylate cyclase; Gαq promotes the synthesis of inositol triphosphate, activates phosphatidylinositol-3 kinase (PI3K), and increases the intracellular concentrations of Ca
++. Gαo activates the Wnt-β-catenin signaling pathway, and Gα
12/13 activates the Rho/Rock signaling pathway and regulates cytoskeletal rearrangements [
25,
30,
31,
32,
33]. Through these mechanisms, SP, after binding to NK-1R, regulates the anti-apoptotic cell proliferation and cell migration signaling pathways involved in cancer development [
34].
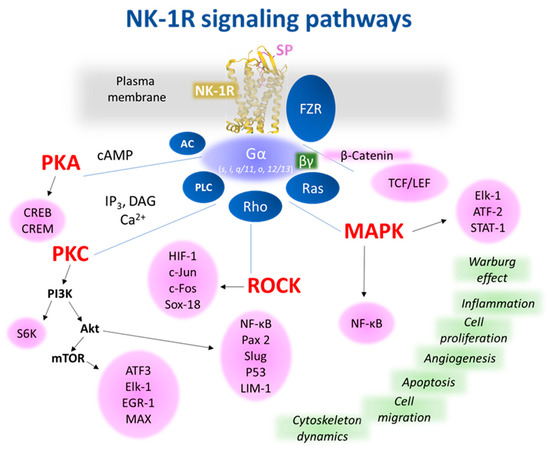
Figure 2 shows the most representative NK-1R downstream pathways involved in cancer progression.
Figure 2. Representative NK-1R intracellular signaling pathways, which are possibly involved in cancer-associated processes. The receptor figure in yellow corresponding to PDB ID 7RMH [
35] is from the Protein Data Bank [
36] drawn with Mol* web-based open-source toolkit [
37]. The transcription factors responsible for controlling cellular events (green) are in pink circles. Abbreviations: AC, adenylyl cyclase; Akt, Ak strain transforming, a protein kinase; ATF, activation transcription factor; cAMP, cyclic adenosine 5′monophosphate; CREB, cAMP response element-binding; CREM, cAMP responsive element modulator; c-Fos, transcription factor; c-Jun, transcription factor; DAG, diacylglycerol; EGR-1, early growth response; Elk-1, ETS-like protein; FZR, Frizzled receptor; HIF, hypoxia-Inducible Factor; IP3, inositol 1,4,5 trisphosphate; LIM-1, Lin11, Isl-1 y Mec-3; MAPK, mitogen-activated protein kinase; MAX, myc-associated factor X; mTOR, mammalian/mechanistic target of rapamycin, a protein kinase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; p53, tumor suppressor protein; PAX-2, paired box gene 2; PI3K, phosphatidylinositol 3 kinase; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; Ras, rat sarcoma virus, a small GTPase; Rho/ROCK, Ras homologous/Rho-associated protein kinase; Slug, SNAI2, a zinc-finger transcription factor; Sox18, SRY-related HMG-box; S6K, ribosomal protein S6 kinase; TCF/LEF, T cell factor/lymphoid enhancer factor family.
2.2. Substance P and Neurokinin-1 Receptor Antagonists
NK-1R shows a preferential affinity for SP and hemokinin-1 (a peripheral ligand of NK-1R), which belong to the tachykinin peptide family [
17]. The affinity of the NK-1R for other members of the tachykinin peptide family, such as neurokinin A and B, is 100- and 500-fold lower than for SP. For this reason, NK-1R is also named the SP receptor [
18]. SP and hemokinin-1 show a high and similar affinity for NK-1R. Both tachykinins share a homologous C-terminal sequence, favor angiogenesis, exert an anti-apoptotic effect, promote the proliferation and migration of tumor cells (e.g., hemokinin-1 increases the expression of matrix metalloproteinases, favors phosphorylation by protein kinase B and extracellular signal-regulated kinases (ERK), and enhances the action of the nuclear factor kappa-light-chain-enhancer of activated B cells) [
18,
38,
39,
40]. Accordingly, NK-1R antagonists inhibit the physiological actions mediated through the NK-1R by SP and hemokinin-1 [
41]. SP is an undecapeptide derived from the pre-protachykinin A gene (the
TAC1 gene locates on chromosome 7 in humans); it is hydrolyzed by p-endopeptidase (extracellular fluid) and angiotensin-converting enzyme (plasma), and it has a half-life from seconds/minutes (tissues) to hours (plasma) [
25]. The undecapeptide is widely distributed by the whole body (e.g., immune and endothelial cells, smooth muscle, nervous system, fibroblasts, platelets, cerebrospinal fluid, blood) and, after binding to NK-1R, it is involved in many physiological and pathophysiological actions: micturition, chemotaxis of leukocytes, sensory perception, cardiovascular and respiratory mechanisms, salivation, movement control, immunological processes, sperm cell motility, platelet aggregation, cellular shape change, neuronal degeneration, memory, neurotransmitters release (e.g., acetylcholine, glutamate, histamine, dopamine), permeability of the blood–brain barrier, gastric motility, nausea and vomiting, obesity, pain, inflammation, anxiety, depression, bipolar disorder, pulpitis, neurocysticercosis, thrombosis, Hirschsprung’s and Crohn’s diseases, heart failure, myocarditis, cholestasis, emesis, migraine, mycosis, urinary incontinence, hepatitis, seizure, pruritus, dermatitis, acute pancreatitis, epilepsy, aggressive behavior, rheumatoid arthritis, asthma, chronic bronchitis, ulcerative colitis, viral and bacterial infection, alcohol addiction, and cancer [
25,
42,
43]. The SP/NK-1R system is upregulated in many pathologies (e.g., inflammation, asthma, virus infection, acute pancreatitis, chronic stress, major depressive disorder, ulcerative colitis, Crohn’s disease, and cancer) [
25]. Patents using NK-1R antagonists against corneal neovascularization, ocular pain, melanogenesis, cough, bacterial infection, respiratory tract diseases, cardiomyopathy, pruritus, emesis, and cancer have been reported [
43]. Despite the potential therapeutic actions of NK-1R antagonists to treat many human pathologies, its potential is currently minimized. It is important to remark that many of the beneficial actions of NK-1R antagonists seen in preclinical studies have been ineffective in clinical trials [
42,
44]. This observation could be due to a lack of knowledge regarding the molecular interactions between NK-1R and NK-1R antagonists, the non-appropriate selection of patients/endpoints in the clinical trials, the non-appropriate use of experimental animal models, and the lower dose of NK-1R antagonists administered. Thus, studies focused on new NK-1R antagonists showing improved pharmacokinetic characteristics are crucial, and must urgently be performed. In addition, for each pathology, the correct dose of the NK-1R antagonist must be administered because, for example, in clinical trials, aprepitant either exerted (300 mg/day for 45 days) or did not exert (160 mg/day for 5–6 days) an antidepressant action, depending on the dose administered [
45,
46]. Thus, the lower dose did not reach the threshold required to promote an antidepressant activity, which could be due to the number of NK-1Rs that must be blocked with aprepitant; it seems that the appropriate dose links to the number of NK-1Rs expressed in cancer cells.
2.3. Neurokinin-1 Receptor Isoforms and Cancer
The expression levels of NK-1R are elevated in cancerous samples obtained from the gallbladder [
47], pancreas [
48], metastatic breast cells [
49], acute myeloid leukemia blasts [
50], esophageal squamous cell carcinoma [
51], or lung cancer cells [
52,
53]. In contrast, neighboring non-affected tissues showed regular expression. Two isoforms of NK-1R have been reported: full-length (407 amino acids) and truncated (311 amino acids, the last 96 residues at the C-terminus are lost) forms [
54]. In some breast cancer cells, the complete structure of NK-1R diminished, and the truncated variant was more abundant [
55]. The two variants may be involved in segregated expressions, responses to ligand concentrations, and triggering different intracellular signaling mechanisms [
56]. The implication of NK-1R increased activity through both receptor forms in cancer cells is supported by the fact that specific antagonists reverse the system’s participation in cell proliferation, migration, and metastasis [
9]. Although precise mechanisms expect determination, there seems to be a dysregulated transcription, where different transcription factors may intervene. Indirect experimental evidence indicates that a putative regulator is a nuclear factor kappa B (NF-κB). This transcription factor governs the expression of NK-1R in macrophage cells by a process dependent on cytokine 1L-1β [
57]. In addition, a pro-inflammatory cytokine cocktail (interferon-γ, tumor necrosis factor-α, and 1L-1β) provoked overexpression of NK-1R in colonic epithelial cells and colonic epithelial cell lines [
58]. However, the order in which molecular events occur inside the cells requires extensive analysis [
56]. Still, the data support that this transcription factor plays a significant role in the hyperactivation of the NK-1R system related to cancer [
59]. The short variant of NK-1R may control the immune environment that could suffer alterations at high concentrations of the ligand SP [
60]. The oncogenic isoform of NK-1R is the truncated form that mediates tumor growth and malignancy. In contrast, the full-length form interacts with β-arrestin, which is involved in the desensitization, internalization, and endocytosis of NK-1R [
18]. The expression of NK-1R mRNA is lower in benign tissues than in malignant ones. Compared with fibroblasts, the expression of the
TACR1 gene was augmented 7.5–30 times in human hepatoblastoma cell lines [
48,
61,
62]. The truncated form is dominantly expressed in cervical and prostate cancer cell lines [
11,
63]. It seems that the truncated form prolongs the response of ligands, because its internalization and desensitization are affected (the absence of amino acid sequences at the C-terminus of NK-1R could block the internalization of the receptor, a clathrin-dependent mechanism, and the recycling processes, leading to SP longer response and cancer progression). In addition, human hepatoblastoma cell lines overexpress the truncated form. Still, negligible levels of this form were found in nonmalignant HEK-293 cells and human fibroblasts [
61,
64,
65,
66]. An increase in SP/NK-1R staining has been reported in metastatic tumors. In cultured normal epithelial cells, the level of SP was lower than that found in cultured cancer cells [
62,
67]. SP, via NK-1R, favors metastasis in human colorectal cancer cells [
68]. The expression of NK-1R isoforms is important since activating the truncated form increases metastasis, whereas the activation of the full-length decreases it [
55]. Moreover, the activation of the full-length isoform inhibited the proliferation of tumor cells, whereas the activation of the truncated form promoted its proliferation [
55]. The tumor growth factor β regulates the expression of the truncated form, an action counteracted by the NK-1R antagonist aprepitant [
69]. Pancreatic ductal adenocarcinoma cells mainly expressed the truncated form. Aprepitant exerted its highest antitumor action against them when tumor cells expressed higher levels of this isoform; in this study, the authors highlighted that NK-1R could be an important target in future personalized medicine [
70]. Moreover, the latter study also reported that the expression of NK-1R was lower in pancreatic ductal adenocarcinoma tissues than in normal tissues. A better overall survival rate was observed in patients showing a high level of NK-1R [
70]. These are unexpected and contradictory findings compared with most of the previously published results; these findings may occur exclusively in pancreatic ductal adenocarcinomas [
9,
70]. This must be elucidated in future studies. A review focused on the structural dynamics and signaling cascades (e.g., mitogen-activated protein kinases (MAPK), hairy and enhancer of split 1 (Hes1)) of NK-1R has recently been published [
71]. For example, Hes 1, a transcriptional blocker of the Notch signaling pathway, reduced the growth suppression of tumor cells when NK-1R was downregulated [
72].
2.4. The Substance P/Neurokinin-1 Receptor System and Cancer: Key Points
The SP/NK-1R system is involved in the molecular bases of many human pathologies, including cancer. This fact means that in-depth knowledge of this system is a crucial step toward better understanding and management of cancer. In the last few years, this knowledge has dramatically increased. Currently, the known key points regarding the involvement of the SP/NK-1R system in cancer are the following: (1) cancer cells express/overexpress NK-1R (e.g., 40,000–60,000 NK-1Rs per glioma cell); (2) NK-1R is involved in the viability of tumor cells (e.g., acute lymphoblastic leukemia, melanoma, lung cancer); (3) NK-1R is not involved in the viability of normal cells; (4) tumor cells express a higher level of truncated NK-1R than normal cells; (5) tumor cells express a lower level of full-length NK-1R than normal cells; (6) SP is not relevant for the viability of cancer cells; (7) SP, after binding to NK-1R (activates MAPK cascade), promotes the mitogenesis/migration of cancer cells (solid and non-solid tumors); (8) cancer cells synthesize and release SP; (9) the undecapeptide acts through endocrine (from tumor mass), paracrine and autocrine mechanisms; (10) SP comes from multiple sources: cancer cells, immune cells placed in the tumor microenvironment, nerve terminals, and bloodstream; (11) SP increases the expression of NK-1R but not that of other tachykinin receptors (NK-2R and NK-3R); (12) SP exerts an anti-apoptotic effect (activating the basal kinase activity of the anti-apoptotic molecule protein kinase B (Akt)) and increases the glycolytic rate (Warburg effect) of tumor cells (which augment their metabolism due to the glucose obtained); (13) SP promotes the growth of blood vessels (angiogenesis, favoring the development of tumors by increasing the supply of blood; endothelial cells express NK-1R and SP), and (14) a higher serum SP level and a higher number of NK-1Rs have been observed in patients with cancer than in healthy individuals [
1,
9,
15,
25,
73,
74,
75,
76,
77,
78,
79,
80,
81,
82,
83,
84,
85,
86]. Moreover, the
TAC and
TACR1 genes are expressed in primary stem cells derived from human placental cord blood and human stem cell lines. SP promoted the proliferation and migration of stem cells; in the latter cells, SP activated the MAPK cascade via NK-1R, and NK-1R antagonists exerted an antitumor activity against cancer stem cells [
87,
88,
89,
90].
2.5. The Substance P/Neurokinin-1 Receptor System as a Cancer Predictive Factor
The overexpression of NK-1R by cancer cells can be used as a prognostic biomarker. It could also facilitate a specific antitumor treatment using NK-1R antagonists (e.g., aprepitant, CP-96,345, L-733,060, SR-140,333, L-732,138). In addition, an increased serum SP level can be used as a predictive factor, indicating a high risk of developing cancer [
9,
15,
91]. The higher level of NK-1R in tumor cells has been related to cancer stage, tumor-node metastasis, poor prognosis, larger tumor size, and higher metastatic and invasion potential [
1,
72,
73,
75,
92,
93]. The expression of NK-1R has been associated with poor prognosis and advanced clinical stages of lung cancer [
14]. A poor prognosis is associated with a high expression of truncated NK-1 in breast cancer, and the expression of SP/NK-1R has been suggested as a predictor for colorectal cancer [
69,
94]. NK-1R has been suggested as a tumor biomarker in hepatoblastoma, independent of the tumor biology and clinical stage. In non-tumor controls, the expression of the truncated NK-1R was lower than that reported in children with hepatoblastoma [
2,
95]. Tumor size and lymph-node metastasis relate to the number of fibers containing SP [
96,
97,
98]. In breast cancer, the overexpression of SP is associated with a negative prognostic value, the expression of NK-1R with a high Ki-67 index (the nuclear Ki-67 protein is related to proliferative mechanisms), and higher tumor grade [
99,
100]. The data suggest that NK-1R and SP may serve as predictive cancer factors.
2.6. The Neurokinin-1 Receptor Is Crucial for the Viability of Cancer Cells
Another significant point is that the expression of the
TACR1 gene is essential for the survival of tumor cells but not for the viability of normal cells; this means that NK-1R is a promising and specific therapeutic target for cancer treatment [
15]. Because the stimulus mediated by SP is beneficial for the survival of cancer cells (e.g., proliferation, migration, anti-apoptotic effect, NK-1R synthesis increase), these cells overexpress NK-1R, ensuring SP binding. Blocking the stimulus with NK-1R antagonists or silencing the expression of the receptor, tumor cells suffer apoptotic mechanisms [
78,
101,
102,
103]. Antibodies against SP promoted apoptosis and decreased epidermal growth factor receptor (EGFR) phosphorylation, as well as the survival of tumor cells. An increase in EGFR expression is associated with an increased expression of SP and a worse prognosis [
104,
105]. When tumor cells do not receive the stimulus mediated by SP, several mechanisms occur: the synthesis of cell cycle proteins halts, the number of apoptotic cells and endothelial growth factor receptors increases, and the steady state of human epidermal growth factor receptor 2 (HER-2) and the MAPK signaling pathway decrease [
104,
105]. NK-1R overexpression could render cancer cells extremely dependent on the SP stimulus, which could counteract the death-signal pathways of tumor cells. This overexpression could neutralize such pathways because death signals can be overridden by the strong SP mitotic stimulus. When blocking NK-1R using NK-1R antagonists, the balance can favor apoptotic signals, leading to cellular death. It has recently been reported that the absence of NK-1R in glioma cells promoted the death of these cells by both apoptotic and necrotic mechanisms. An irreversible lesion, derived from a non-physiological situation, favors the breakage of glioma cell membranes, promoting their death by necrotic mechanisms [
15]. This observation is another vital point worth studying in depth. Altogether, these findings demonstrate the crucial importance of the activation of NK-1R by SP for the survival of cancer cells.
2.7. Neurokinin-1 Receptor and EGFR, Akt, and HER-2
The activation of NK-1R by SP promotes EGFR transactivation, facilitates the formation of EGFR complex, activates the extracellular signal-regulated kinase (ERK) 2 and MAPK pathway, and induces DNA synthesis (
Figure 2) [
106]. ERK can be translocated into the nucleus, promoting proliferation and exerting an anti-apoptotic action. By activating ERK1/2, SP induced the proliferation and migration of cancer cells, and β-arrestin increased the sensitivity of cancer cells to NK-1R antagonists [
107,
108]. EGFR inhibitors blocked DNA synthesis and ERK2 activation mediated by SP, and EGFR transactivation mediated the mitogenic action promoted by SP [
106]. EGFR and c-Src interact, and an augmented activity of c-Src has been associated with cancer progression, as this interaction enhances mitogenic signaling pathways [
109]. Src kinase inhibitors block SP-dependent ERK phosphorylation and suppress the growth of tumor cells [
110]. NK-1R, via EGFR transactivation, promoted non-small cell lung cancer progression (cell proliferation and migration) and aprepitant increased the sensitivity of lung cancer cells to gefitinib or osimertinib [
111]. SP, via PI3K, augments the activity of protein kinase B (Akt), suppressing apoptosis. The inhibition of PI3K increased apoptosis and decreased cellular proliferation in cancer cells [
29,
79,
112,
113,
114]. Akt activation has been related to poor prognosis and cellular processes that avoid the death of cells, leading to drug resistance and decreasing the antitumor effect of aprepitant [
2,
115,
116]. Moreover, SP activates HER-2, which promotes drug resistance and malignant progression [
104,
117].
2.8. Neurokinin-1 Receptor and the Warburg Effect
SP, through NK-1R, mediates the Warburg effect (in tumor cells, compared with normal cells, the glycolytic rate is 200 times higher). This effect is blocked with NK-1R antagonists; then, tumor cells die by starvation (anti-Warburg effect) because NK-1R is needed to obtain glucose (
Figure 2) [
78,
118]. SP activates glycogen synthase kinase-3 (GSK-3β), a finding associated with cancer progression and poor prognosis; its inhibition blocked tumorigenesis, counteracted the Warburg effect, increased apoptosis, and restrained cell motility [
119,
120]. NK-1R antagonists block GSK-3β activity and increase glycogen synthesis, counteracting the Warburg effect [
117].
2.9. Neurokinin-1 Receptor Isoforms Balance
Cancer cells are highly responsive to NK-1R antagonists when they express a high quantity of truncated NK-1R. The overexpression of this form has been associated with an enhanced malignant potential, and the truncated form promoted the malignant transformation of non-tumorigenic cells. Cancer cells express more truncated than full-length forms, and the expression of the full-length form is inversely related to the proliferation of tumor cells, invasiveness, and metastasis [
53,
55,
72,
80,
121]. The truncated form facilitated the synthesis of cytokines favoring growth-promoting actions, and the overexpression of miR-206 by tumor cells favored the malignant phenotype of these cells by maintaining a low level of the full-length isoform [
92,
122]. Thus, the antitumor effect mediated by NK-1R antagonists is associated with the differential expression of NK-1R isoforms; this is a significant point [
2,
34]. Accordingly, knowing the total number of full-length and truncated NK-1R isoforms expressed in tumors is necessary for the in-depth understanding of the mechanisms by which the SP/NK-1R system is involved in cancer, as well as the antitumor actions mediated by NK-1R antagonists.
2.10. Neurokinin-1 Receptor and Inflammation
SP enhanced the inflammatory-mediated tumor signaling pathways and promoted the expression of genes that facilitate tumor growth, invasion, and metastasis in head and neck cancers; these mechanisms were blocked with the NK-1R antagonist L-703,606 [
123]. The SP/NK-1R system has been involved in the transition from chronic inflammation of the neck and head mucosa to preneoplastic/neoplastic transformation and development [
111]. SP activates pro-inflammatory transcriptions factors (e.g., NF-κB) that control the expression of cytokines; NF-κB promotes the synthesis and release of pro-inflammatory cytokines (e.g., tumor necrosis factor; interleukins 1, 6, and 12), and an NF-κB binding site has been located in the promoter of the
TACR1 gene [
25,
124]. SP increases the release of cytokines by monocytes and macrophages, and cancer cells synthesize interleukin 6, being that its level is related to an increased progression of tumors; in addition, this interleukin has been located in the tumor cyst and cerebrospinal fluids of patients with glioma [
124,
125,
126,
127]. Previous data highlight the crucial roles played by SP on inflammatory mechanisms and tumor microenvironment: SP acts as a pro-inflammatory agent, and crosstalk between immune and tumor cells occurs. Chronic inflammation is associated with an increased risk of cancer development and truncated NK-1R/protein levels were higher in colonic epithelial cells with high-grade dysplasia and carcinoma than in quiescent colitis [
128,
129].