Within the 2,5-dioxopiperazine-containing natural products generated by “head-to-tail” cyclization of peptides, those derived from tryptophan allow further structural diversification due to the rich chemical reactivity of the indole heterocycle, which can generate tetracyclic fragments of hexahydropyrrolo[2,3-b]indole or pyrrolidinoindoline skeleton fused to the 2,5-dioxopiperazine. Even more complex are the dimeric bispyrrolidinoindoline epi(poly)thiodioxopiperazines (BPI-ETPs), since they feature transannular (poly)sulfide bridges connecting C3 and C6 of their 2,5-dioxopiperazine rings. Homo- and heterodimers composed of diastereomeric epi(poly)thiodioxopiperazines increase the complexity of the family.
1. Introduction
Dimeric bispyrrolidinoindoline epi(poly)thiodioxopiperazines (BPI-ETPs) are a family of highly complex natural products that biogenetically derive from dioxopiperazines formed by the double intramolecular condensation of dipeptides containing tryptophan and an additional amino acid, followed by a variety of structural modifications [
1,
2,
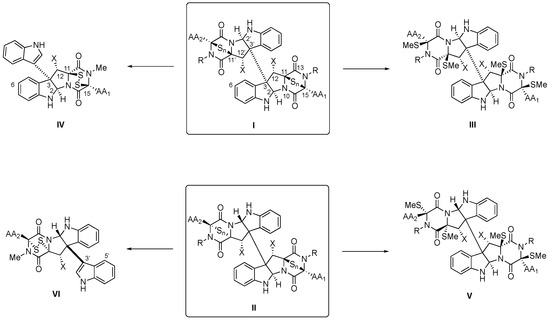
3]. The monomeric units contain cyclic dipeptide (CDP) substructures fused to pyrrolidinoindoline cores and feature transannular (poly)sulfide connections between C3 and C6 (for the numbering indicated in
Scheme 1, see [
4]) of their 2,5-dioxopiperazine rings [
1]. Relevant features of these privileged structures include the conformational constraint, which has been linked to their greater stability and conformational rigidity and, therefore, higher resistance to protease degradation than acyclic counterparts, as well as their ability to cross the intestinal barrier and the blood–brain barrier [
5]. The ability to mimic preferential peptide conformations, with two hydrogen bond donor and acceptor sites, favors interactions with putative biological targets. Thus, their pharmacological potency is boosted when compared with the monomeric unit through multipoint interactions on chemical space with their biological targets [
6,
7].
Scheme 1. General structures (and atom numbers) of tryptophan-derived BPI-ETP alkaloids (I,II), their transformation into the methylsulfanyl derivatives (III,V), and partial degradation products (IV,VI).
More than 100 alkaloids containing at least a unit of indole-derived [
8] pyrrolidinoindoline epi(poly)thiodioxopiperazine (PI-ETP) [
9] have already been isolated from terrestrial and marine fungi (about a third, from marine sources) [
10,
11,
12,
13], including species of the
Leptosphaeria,
Chaetomium, [
14]
Tilachlidium,
Verticillium,
Gliocladium,
Aspergillus sp. and
Penicillium genera [
1,
15,
16], but they have also been obtained from bacteria (
Streptomyces species) [
17], plants, and even animals [
5,
13,
18]. Homo- and heterodimeric BPI ETPs compose about half of the family.
(+)-Chaetocin A (
1, Figure 1A) [
19] and (+)-verticillin A (
8, Figure 2) [
20] were first discovered in 1970, although antibacterial monomeric (−)-gliotoxin (
7, see Figure 1B) had been isolated more than 30 years before [
21,
22]. Relative to the monomers, the homo- and heterodimeric BPI ETP family members feature a greater level of structural diversity and complexity since the characteristic dioxopiperazine scaffold is bridged by a variable number (1 to 4) of sulfur atoms. Nondimeric family members connected at C3 through the indole nitrogen of either simple indole units or epi(poly)thiodioxopiperazines will not be covered.
Biogenetic generation of CDPs in nature usually involves either nonribosomal peptide synthetases (NRPSs) in fungi [
23,
24] or cyclodipeptide synthases (CDPSs) in bacteria [
25,
26,
27]. Whereas fungi generate DKP monomers by the activation of the free amino acids through adenylation by the action of NRPSs, bacteria use cyclodipeptide synthetases (CDPSs) and employ aminoacyl-tRNAs (aa-TRNAs) as substrates [
26,
27,
28,
29,
30].
The diversification of the skeleton is further achieved through the biogenetically controlled action of their tailoring enzymes, which are usually found in dedicated biosynthetic gene clusters [
31]. Whereas, for the bispyrrolidinoindoline dioxopiperazine, family various types of oxidoreductases, hydrolases, transferases, and ligases have been isolated and structurally characterized [
32], for the BPI-ETP family, these chemical modifications are rather limited. In addition to the role of Cyt P450 oxidation enzymes [
33], as well as enzymatic dimerization [
34,
35], only hydroxylation/dehydroxylation processes leading ultimately to the modulation of the oxidation level of the pyrrolidinoindoline and DKP scaffolds and the amino acid side chains have been noted in some members of the family of dimeric alkaloids. In contrast, enzymes responsible for structural modifications of the DKP scaffold in simpler bispyrrolidinoindoline dioxopiperazine alkaloids have been characterized in several biogenetic gene clusters of fungal and microbial secondary metabolites [
1,
5,
29,
36,
37,
38,
39,
40,
41,
42].
The conformationally constrained DKP scaffold of dimeric dioxopiperazines is currently considered as privileged structures [
43], given their ability to interact with several receptors, which may account for the diverse biological activities reported for this family of natural products. In addition, the reactivity of compounds with di(poli)sulfide bridges has been associated with a variety of biological activities, including protein cross-linking through the reaction of the disulfide bond with cysteine residues and the inactivation of thiol-containing proteins, generation of reactive oxygen species (ROS) via redox cycling, or ejection of zinc ions from some proteins [
10,
11,
12,
44,
45,
46].
2. Biogenesis of BPI-ETPs
Biogenetic studies of sulfur containing moieties in natural products [
105], in particular those from marine organisms [
106], as well as comprehensive report on the biosynthesis of pyrrolidinoindoline containing NPs, including BPI-ETPs [
107], together with the flavoenzyme-catalyzed formation of disulfide bonds in natural products biosynthesis [
108,
109], have been previously covered. The biogenetic routes to these natural products are usually found in dedicated biosynthetic gene clusters [
31,
110,
111].
Whereas the incorporation of sulfur to a putative dehydrodioxopiperazine was first proposed to occur immediately following the cyclodipeptide formation,
N-methylation was proposed to occur after the oxidative cyclization [
112]. Methionine, cysteine, and sodium sulfate jointly provide the sulfur connections of thiodioxopiperazines [
10].
Most of the biosynthetic studies have been carried out on monomeric gliotoxin (
7,
Figure 1B) from
Aspergillus fumigatus, in which the dioxopiperazine core is assembled by a nonribosomal peptide synthetase [
113]. The presence in a fermentation broth of a cyclic dipeptide intermediate bound to glutathione suggested the latter to be the donor of sulfur atoms [
114]. Gene knockout experiments revealed that
gliG was responsible for encoding a glutathione sulfur transferase, namely, GliG, which incorporated the sulfur atom into the DKP framework. Prior to sulfur incorporation, the DKP should undergo an oxidation with the attachment of a hydroxyl group at the Cα-position, for which gliC was identified as the responsible P450 monooxygenase [
46,
113,
114].
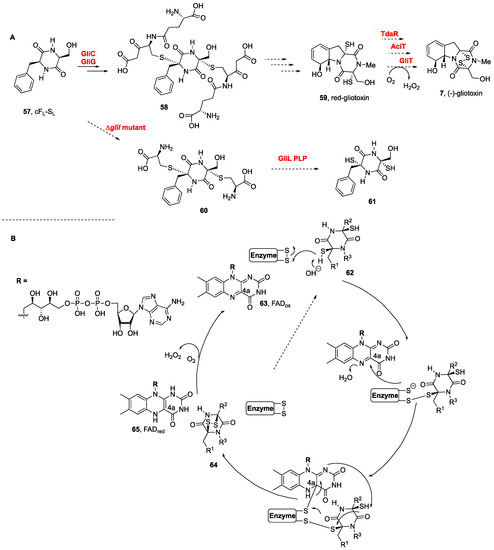
The biosynthesis of (−)-gliotoxin (
7,
Scheme 2A) in
Aspergillus fumigatus was elucidated by Hertweck et al., after their discovery of the activation of DKPs by oxygenase GliC and the transfer of glutathione by a dedicated glutathione
S-transferase, GliC [
114,
115], which led to the formation of bis(glutathione) adducts
58 from cF
L-S
L (
57) and lately to the natural product from the dithiol intermediate
59 (
Scheme 2) [
114]. Additional insights were obtained from the large-scale fermentation of an engineered ΔgliI mutant, from which bis(cysteine) conjugates (
60) were indeed isolated, en route to the dithiol intermediate
61. The step involving the dual cleavage of the C–S bonds to afford the dithiol prior to disulfide bond formation was also confirmed. The GliI C–S lyase-catalyzed reaction concomitantly forms two thiol groups, in what might reflect the dual action of the GliI homodimer [
116].
Scheme 2. (
A) Biogenetic route to pyrrolidinoindoline epidithiodioxopiperazine (−)-gliotoxin (
7) [
114]. (
B) Proposal for enzymatic disulfide bond formation [
35].
More recent studies on the biogenesis of pretrichodermamide A (not shown) containing a formal α,β-disulfide (instead of the α,α-disulfide of this series) in
Trichoderma hypoxylon confirmed that the formation of the disulfide bond to generate the epidithiodioxopiperazine was promoted by an FAD (
63)-dependent oxidoreductase (
Scheme 2B). The thiol disulfide oxidases promoted the disulfide bond formation from dithiol
62 to disulfide
64 with substrate and catalytic promiscuities. Not only the disulfide bond of pretrichodermamide A (not shown) (TdaR) but also those of aspirochlorine (not shown) (AclT) and gliotoxin (
7,
Figure 1) (GliT) were generated efficiently (see
Scheme 2B) [
35].
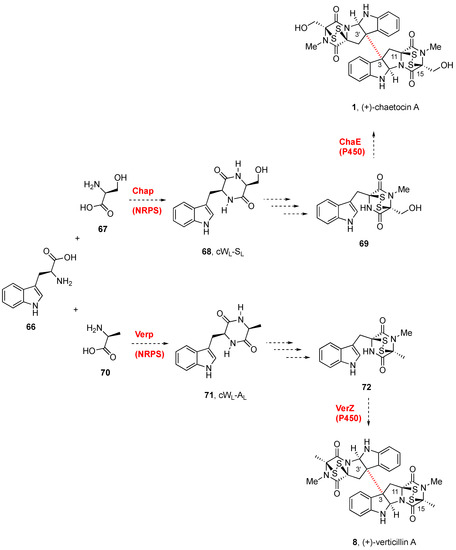
The biosynthetic gene cluster (
cha) of (+)-chaetocin A (
1,
Figure 1) was identified in the producing fungi
Chaetomium virescens ATCC 26417 through bioinformatic comparison with the BGCs of (−)-gliotoxin (
7,
Figure 1) and sirodesmin (not shown) [
117]. Three cytochrome P450 enzymes (ChaB, ChaC, and ChaE) were identified as part of the cha cluster. Although the gene cluster
cha from
Chaetomium virescens ATCC 26417 was proposed to generate (+)-chaetocin A (
1,
Scheme 3) [
117], through radical dimerization, it has not been possible to express ChaE and confirm its activity. Given that detailed information on the tailoring of the glutathione adduct by the γ-glutamylcyclotransferase to generate the DKP disulfide, the timing of sulfuration, the DKP release, and the dimerization, has not yet been clarified, the biogenesis of these natural compounds is still uncertain [
117].
Scheme 3. Biogenetic routes to BPI-ETP alkaloids.
The discovery of the gene cluster for the biosynthesis of (+)-verticillin A (
8,
Scheme 3) [
118] confirmed that
verP encoded a NPRS in connection with other genes besides
verA and
verK. The disulfide bond formation by oxidation of the dithiols was likely promoted by VerT, similar to GliT, and
verZ controls the production of the natural product [
119]. The biogenesis of (+)-verticillin A (
8) was abolished by the disruption of
verM or
verG, encoding a putative
O-methyltransferase and a glutathione
S-methyltransferase, respectively [
118,
119]. These mutants produced unrelated metabolites termed gliocladiosin A and B (structures not shown). No additional mechanistic details have been reported for the series.
The biosynthetic gene cluster of (+)-verticillin A (
8) from the
Cordyceps-colonizing fungus
Clonostachys rogersoniana (formerly known as
Gliocladium sp.) isolated from the
Cordycep fruiting body collected in Tibet Linzhi County (China) has more recently been identified and cloned (
Scheme 3). Based on the sequence of a nonribosomal peptide synthetase (ChaP), which was predicted to be responsible for (+)-chaetocin A (
1) biosynthesis in
C. virescens, the verticillin biosynthetic gene cluster (
ver) was constructed. Disruption of the nonribosomal peptide synthetase gene
verP in the
ver cluster was shown to abolish the production of (+)-verticillin A (
8) [
120]. Disruption of the
O-methyltransferase gene
verM on the same species stopped the production of (+)-verticillin A (
8) and allowed for isolating two cryptic compounds (not shown), which were identified as dipeptides conjugated with macrolides [
121].
This entry is adapted from the peer-reviewed paper 10.3390/molecules27217585