Prions are misfolded proteins that have the ability to transmit their misfolded shape onto normal variants of the same protein. They characterize several fatal and transmissible neurodegenerative diseases in humans and many other animals. It is not known what causes a normal protein to misfold, but the resulting abnormal three-dimensional structure confers infectious properties by collapsing nearby protein molecules into the same shape. The word prion is derived from the term, "proteinaceous infectious particle". In comparison to all other known infectious agents such as viroids, viruses, bacteria, fungi, and parasites, all of which contain nucleic acids (DNA, RNA, or both), the hypothesized role of a protein as an infectious agent stands in contrast. Prion isoforms of the prion protein (PrP), whose specific function is uncertain, are hypothesized as the cause of transmissible spongiform encephalopathies (TSEs), including scrapie in sheep, chronic wasting disease (CWD) in deer, bovine spongiform encephalopathy (BSE) in cattle (commonly known as "mad cow disease") and Creutzfeldt–Jakob disease (CJD) in humans. All known prion diseases in mammals affect the structure of the brain or other neural tissue; all are progressive, have no known effective treatment, and are always fatal. Until 2015, all known mammalian prion diseases were caused by the prion protein (PrP); however, in 2015 it was hypothesized that multiple system atrophy (MSA) was caused by a prion form of alpha-synuclein. Prions are a type of intrinsically disordered protein, which change their conformation unless they are bound to a specific partner such as another protein. With a prion, two protein chains are stabilized if one binds to another in the same conformation. The probability of this happening is low, but once it does the combination of the two is very stable. Then more units can get added, making a sort of "fibril". Prions form abnormal aggregates of proteins called amyloids, which accumulate in infected tissue and are associated with tissue damage and cell death. Amyloids are also responsible for several other neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease. A prion disease is a type of proteopathy, or disease of structurally abnormal proteins. In humans, prions are believed to be the cause of Creutzfeldt–Jakob disease (CJD), its variant (vCJD), Gerstmann–Sträussler–Scheinker syndrome (GSS), fatal familial insomnia (FFI), and kuru. There is also evidence suggesting prions may play a part in the process of Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS); these have been termed prion-like diseases. Several yeast proteins have also been identified as having prionogenic properties, as well as a protein involved in modification of synapses during the formation of memories (see Eric Kandel § Molecular changes during learning). Prion replication is subject to epimutation and natural selection just as for other forms of replication, and their structure varies slightly between species. Prion aggregates are stable, and this structural stability means that prions are resistant to denaturation by chemical and physical agents: they cannot be destroyed by ordinary disinfection or cooking. This makes disposal and containment of these particles difficult.
- familial
- structural stability
- encephalopathy
1. Etymology and Pronunciation
The word prion, coined in 1982 by Stanley B. Prusiner, is derived from protein and infection, hence prion,[1][2] and is short for "proteinaceous infectious particle",[3] in reference to its ability to self-propagate and transmit its conformation to other proteins.[4] Its main pronunciation is /ˈpriːɒn/ (listen),[5][6][7] although /ˈpraɪɒn/, as the homographic name of the bird (prions or whalebirds) is pronounced,[7] is also heard.[8] In his 1982 paper introducing the term, Prusiner specified that it is "pronounced pree-on".[9]
2. Prion Protein
2.1. Structure
The protein that prions are made of (PrP) is found throughout the body, even in healthy people and animals. However, PrP found in infectious material has a different structure and is resistant to proteases, the enzymes in the body that can normally break down proteins. The normal form of the protein is called PrPC, while the infectious form is called PrPSc – the C refers to 'cellular' PrP, while the Sc refers to 'scrapie', the prototypic prion disease, occurring in sheep.[10] While PrPC is structurally well-defined, PrPSc is certainly polydisperse and defined at a relatively poor level. PrP can be induced to fold into other more-or-less well-defined isoforms in vitro, and their relationship to the form(s) that are pathogenic in vivo is not yet clear.
PrPC
PrPC is a normal protein found on the membranes of cells, "including several blood components of which platelets constitute the largest reservoir in humans."[11] It has 209 amino acids (in humans), one disulfide bond, a molecular mass of 35–36 kDa and a mainly alpha-helical structure. Several topological forms exist; one cell surface form anchored via glycolipid and two transmembrane forms.[12] The normal protein is not sedimentable; meaning that it cannot be separated by centrifuging techniques.[13] Its function is a complex issue that continues to be investigated. PrPC binds copper (II) ions with high affinity.[14] The significance of this finding is not clear, but it is presumed to relate to PrP structure or function. PrPC is readily digested by proteinase K and can be liberated from the cell surface in vitro by the enzyme phosphoinositide phospholipase C (PI-PLC), which cleaves the glycophosphatidylinositol (GPI) glycolipid anchor.[15] PrP has been reported to play important roles in cell-cell adhesion and intracellular signaling in vivo, and may therefore be involved in cell-cell communication in the brain.[16]
PrPres
Protease-resistant PrPSc-like protein (PrPres) is the name given to any isoform of PrPc which is structurally altered and converted into a misfolded proteinase K-resistant form in vitro.[17] To model conversion of PrPC to PrPSc in vitro, Saborio et al. rapidly converted PrPC into a PrPres by a procedure involving cyclic amplification of protein misfolding.[18] The term "PrPres" has been used to distinguish between PrPSc, which is isolated from infectious tissue and associated with the transmissible spongiform encephalopathy agent.[19] For example, unlike PrPSc, PrPres may not necessarily be infectious.
PrPSc
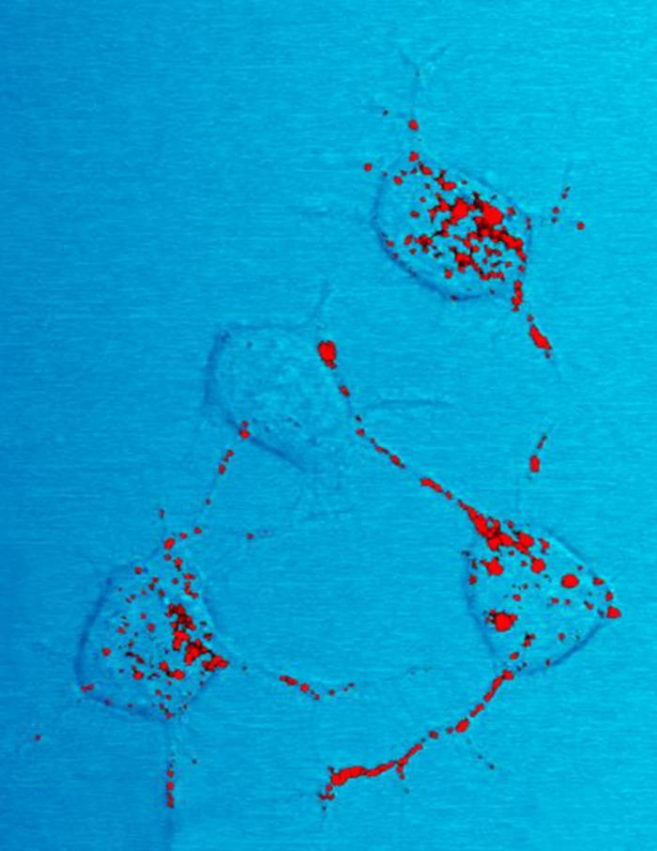
The infectious isoform of PrP, known as PrPSc, or simply the prion, is able to convert normal PrPC proteins into the infectious isoform by changing their conformation, or shape; this, in turn, alters the way the proteins interconnect. PrPSc always causes prion disease. Although the exact 3D structure of PrPSc is not known, it has a higher proportion of β-sheet structure in place of the normal α-helix structure.[20] Aggregations of these abnormal isoforms form highly structured amyloid fibers, which accumulate to form plaques. The end of each fiber acts as a template onto which free protein molecules may attach, allowing the fiber to grow. Under most circumstances, only PrP molecules with an identical amino acid sequence to the infectious PrPSc are incorporated into the growing fiber.[13] However, rare cross-species transmission is also possible.[21]
2.2. Normal Function of PrP
The physiological function of the prion protein remains poorly understood. While data from in vitro experiments suggest many dissimilar roles, studies on PrP knockout mice have provided only limited information because these animals exhibit only minor abnormalities. In research done in mice, it was found that the cleavage of PrP in peripheral nerves causes the activation of myelin repair in Schwann cells and that the lack of PrP proteins caused demyelination in those cells.[22]
PrP and regulated cell death
MAVS, RIP1, and RIP3 are prion-like proteins found in other parts of the body. They also polymerise into filamentous amyloid fibers which initiate regulated cell death in the case of a viral infection to prevent the spread of virions to other, surrounding cells.[23]
PrP and long-term memory
A review of evidence in 2005 suggested that PrP may have a normal function in maintenance of long-term memory.[24] As well, a 2004 study found that mice lacking genes for normal cellular PrP protein show altered hippocampal long-term potentiation.[25][26] A recent study that might explain why this is found that neuronal protein CPEB has a similar genetic sequence to yeast prion proteins. The prion-like formation of CPEB is essential for maintaining long-term synaptic changes associated with long-term memory formation.[27]
PrP and stem cell renewal
A 2006 article from the Whitehead Institute for Biomedical Research indicates that PrP expression on stem cells is necessary for an organism's self-renewal of bone marrow. The study showed that all long-term hematopoietic stem cells express PrP on their cell membrane and that hematopoietic tissues with PrP-null stem cells exhibit increased sensitivity to cell depletion.[28]
PrP and innate immunity
There is some evidence that PrP may play a role in innate immunity, as the expression of PRNP, the PrP gene, is upregulated in many viral infections and PrP has antiviral properties against many viruses, including HIV.[29]
3. Prion Replication

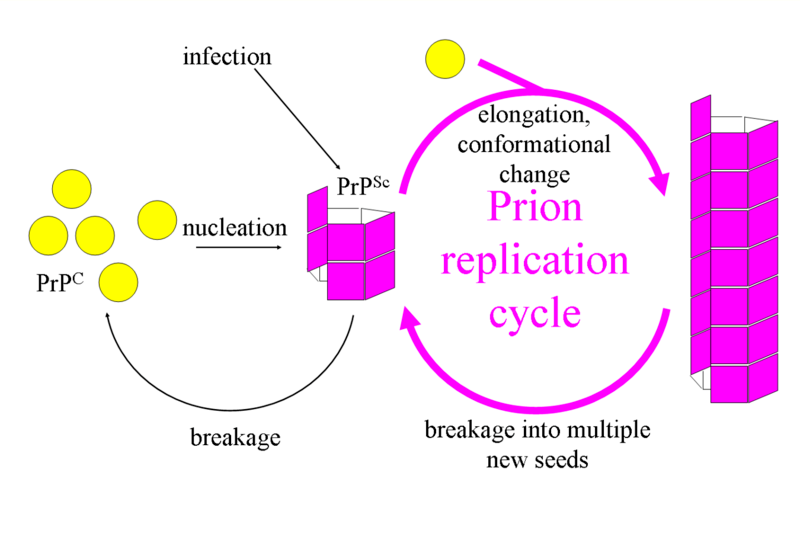
The first hypothesis that tried to explain how prions replicate in a protein-only manner was the heterodimer model.[30] This model assumed that a single PrPSc molecule binds to a single PrPC molecule and catalyzes its conversion into PrPSc. The two PrPSc molecules then come apart and can go on to convert more PrPC. However, a model of prion replication must explain both how prions propagate, and why their spontaneous appearance is so rare. Manfred Eigen showed that the heterodimer model requires PrPSc to be an extraordinarily effective catalyst, increasing the rate of the conversion reaction by a factor of around 1015.[31] This problem does not arise if PrPSc exists only in aggregated forms such as amyloid, where cooperativity may act as a barrier to spontaneous conversion. What is more, despite considerable effort, infectious monomeric PrPSc has never been isolated.
An alternative model assumes that PrPSc exists only as fibrils, and that fibril ends bind PrPC and convert it into PrPSc. If this were all, then the quantity of prions would increase linearly, forming ever longer fibrils. But exponential growth of both PrPSc and of the quantity of infectious particles is observed during prion disease.[32][33][34] This can be explained by taking into account fibril breakage.[35] A mathematical solution for the exponential growth rate resulting from the combination of fibril growth and fibril breakage has been found.[36] The exponential growth rate depends largely on the square root of the PrPC concentration.[36] The incubation period is determined by the exponential growth rate, and in vivo data on prion diseases in transgenic mice match this prediction.[36] The same square root dependence is also seen in vitro in experiments with a variety of different amyloid proteins.[37]
The mechanism of prion replication has implications for designing drugs. Since the incubation period of prion diseases is so long, an effective drug does not need to eliminate all prions, but simply needs to slow down the rate of exponential growth. Models predict that the most effective way to achieve this, using a drug with the lowest possible dose, is to find a drug that binds to fibril ends and blocks them from growing any further.[38]
Researchers at Dartmouth College discovered that endogenous host cofactor molecules such as the phospholipid molecule (e.g. phosphatidylethanolamine) and polyanions (e.g. single stranded RNA molecules) are necessary to form PrPSc molecules with high levels of specific infectivity in vitro, whereas protein-only PrPSc molecules appear to lack significant levels of biological infectivity.[39][40]
4. Transmissible Spongiform Encephalopathies
| Affected animal(s) | Disease |
|---|---|
| Sheep, Goat | Scrapie[41] |
| Cattle | Mad cow disease[41] |
| Camel[42] | Camel spongiform encephalopathy (CSE) |
| Mink[41] | Transmissible mink encephalopathy (TME) |
| White-tailed deer, elk, mule deer, moose[41] | Chronic wasting disease (CWD) |
| Cat[41] | Feline spongiform encephalopathy (FSE) |
| Nyala, Oryx, Greater Kudu[41] | Exotic ungulate encephalopathy (EUE) |
| Ostrich[43] | Spongiform encephalopathy (unknown if transmissible) |
| Human | Creutzfeldt–Jakob disease (CJD)[41] |
| Iatrogenic Creutzfeldt–Jakob disease (iCJD) | |
| Variant Creutzfeldt–Jakob disease (vCJD) | |
| Familial Creutzfeldt–Jakob disease (fCJD) | |
| Sporadic Creutzfeldt–Jakob disease (sCJD) | |
| Gerstmann–Sträussler–Scheinker syndrome (GSS)[41] | |
| Fatal familial insomnia (FFI)[44] | |
| Kuru[41] | |
| Familial spongiform encephalopathy[45] | |
| Variably protease-sensitive prionopathy (VPSPr) |
Prions cause neurodegenerative disease by aggregating extracellularly within the central nervous system to form plaques known as amyloids, which disrupt the normal tissue structure. This disruption is characterized by "holes" in the tissue with resultant spongy architecture due to the vacuole formation in the neurons.[46] Other histological changes include astrogliosis and the absence of an inflammatory reaction.[47] While the incubation period for prion diseases is relatively long (5 to 20 years), once symptoms appear the disease progresses rapidly, leading to brain damage and death.[48] Neurodegenerative symptoms can include convulsions, dementia, ataxia (balance and coordination dysfunction), and behavioural or personality changes.
Many different mammalian species can be affected by prion diseases, as the prion protein (PrP) is very similar in all mammals.[49] Due to small differences in PrP between different species it is unusual for a prion disease to transmit from one species to another. The human prion disease variant Creutzfeldt–Jakob disease, however, is thought to be caused by a prion that typically infects cattle, causing bovine spongiform encephalopathy and is transmitted through infected meat.[50]
All known prion diseases are untreatable and fatal.[51] However, a vaccine developed in mice may provide insight into providing a vaccine to resist prion infections in humans.[52] Additionally, in 2006 scientists announced that they had genetically engineered cattle lacking a necessary gene for prion production – thus theoretically making them immune to BSE,[53] building on research indicating that mice lacking normally occurring prion protein are resistant to infection by scrapie prion protein.[54] In 2013, a study revealed that 1 in 2,000 people in the United Kingdom might harbour the infectious prion protein that causes vCJD.[55]
Until 2015 all known mammalian prion diseases were considered to be caused by the prion protein, PrP; in 2015 multiple system atrophy was found to be transmissible and was hypothesized to be caused by a new prion, the misfolded form of a protein called alpha-synuclein.[3] The endogenous, properly folded form of the prion protein is denoted PrPC (for Common or Cellular), whereas the disease-linked, misfolded form is denoted PrPSc (for Scrapie), after one of the diseases first linked to prions and neurodegeneration.[13][56] The precise structure of the prion is not known, though they can be formed spontaneously by combining PrPC, homopolymeric polyadenylic acid, and lipids in a protein misfolding cyclic amplification (PMCA) reaction even in the absence of pre-existing infectious prions.[39] This result is further evidence that prion replication does not require genetic information.[57]
4.1. Transmission
It has been recognized that prion diseases can arise in three different ways: acquired, familial, or sporadic.[58] It is often assumed that the diseased form directly interacts with the normal form to make it rearrange its structure. One idea, the "Protein X" hypothesis, is that an as-yet unidentified cellular protein (Protein X) enables the conversion of PrPC to PrPSc by bringing a molecule of each of the two together into a complex.[59]
The primary method of infection in animals is through ingestion. It is thought that prions may be deposited in the environment through the remains of dead animals and via urine, saliva, and other body fluids. They may then linger in the soil by binding to clay and other minerals.[60]
A University of California research team has provided evidence for the theory that infection can occur from prions in manure.[61] And, since manure is present in many areas surrounding water reservoirs, as well as used on many crop fields, it raises the possibility of widespread transmission. It was reported in January 2011 that researchers had discovered prions spreading through airborne transmission on aerosol particles, in an animal testing experiment focusing on scrapie infection in laboratory mice.[62] Preliminary evidence supporting the notion that prions can be transmitted through use of urine-derived human menopausal gonadotropin, administered for the treatment of infertility, was published in 2011.[63]
Prions in plants
In 2015, researchers at The University of Texas Health Science Center at Houston found that plants can be a vector for prions. When researchers fed hamsters grass that grew on ground where a deer that died with chronic wasting disease (CWD) was buried, the hamsters became ill with CWD, suggesting that prions can bind to plants, which then take them up into the leaf and stem structure, where they can be eaten by herbivores, thus completing the cycle. It is thus possible that there is a progressively accumulating number of prions in the environment.[64][65]
4.2. Sterilization
Infectious particles possessing nucleic acid are dependent upon it to direct their continued replication. Prions, however, are infectious by their effect on normal versions of the protein. Sterilizing prions, therefore, requires the denaturation of the protein to a state in which the molecule is no longer able to induce the abnormal folding of normal proteins. In general, prions are quite resistant to proteases, heat, ionizing radiation, and formaldehyde treatments,[66] although their infectivity can be reduced by such treatments. Effective prion decontamination relies upon protein hydrolysis or reduction or destruction of protein tertiary structure. Examples include sodium hypochlorite, sodium hydroxide, and strongly acidic detergents such as LpH.[67]
The World Health Organization recommends any of the following three procedures for the sterilization of all heat-resistant surgical instruments to ensure that they are not contaminated with prions:
- Immerse in 1N sodium hydroxide and place in a gravity-displacement autoclave at 121 °C for 30 minutes; clean; rinse in water; and then perform routine sterilization processes.
- Immerse in 1N sodium hypochlorite (20,000 parts per million available chlorine) for 1 hour; transfer instruments to water; heat in a gravity-displacement autoclave at 121 °C for 1 hour; clean; and then perform routine sterilization processes.
- Immerse in 1N sodium hydroxide or sodium hypochlorite (20,000 parts per million available chlorine) for 1 hour; remove and rinse in water, then transfer to an open pan and heat in a gravity-displacement (121 °C) or in a porous-load (134 °C) autoclave for 1 hour; clean; and then perform routine sterilization processes.[68]
134 °C (273 °F) for 18 minutes in a pressurized steam autoclave has been found to be somewhat effective in deactivating the agent of disease.[69][70] Ozone sterilization is currently being studied as a potential method for prion denaturation and deactivation.[71] Other approaches being developed include thiourea-urea treatment, guanidinium chloride treatment,[72] and special heat-resistant subtilisin combined with heat and detergent.[73] A method sufficient for sterilizing prions on one material may fail on another.[74]
Renaturation of a completely denatured prion to infectious status has not yet been achieved; however, partially denatured prions can be renatured to an infective status under certain artificial conditions.[75]
4.3. Degradation Resistance in Nature
Overwhelming evidence shows that prions resist degradation and persist in the environment for years, and proteases do not degrade them. Experimental evidence shows that unbound prions degrade over time, while soil-bound prions remain at stable or increasing levels, suggesting that prions likely accumulate in the environment.[76][77] One 2015 study by US scientists found that repeated drying and wetting may render soil bound prions less infectious, although this was dependent on the soil type they were bound to.[78]
5. Fungi
Proteins showing prion-type behavior are also found in some fungi, which has been useful in helping to understand mammalian prions. Fungal prions do not appear to cause disease in their hosts.[79] In yeast, protein refolding to the prion configuration is assisted by chaperone proteins such as Hsp104.[80] All known prions induce the formation of an amyloid fold, in which the protein polymerises into an aggregate consisting of tightly packed beta sheets. Amyloid aggregates are fibrils, growing at their ends, and replicate when breakage causes two growing ends to become four growing ends. The incubation period of prion diseases is determined by the exponential growth rate associated with prion replication, which is a balance between the linear growth and the breakage of aggregates.[36]
Fungal proteins exhibiting templated conformational change were discovered in the yeast Saccharomyces cerevisiae by Reed Wickner in the early 1990s. For their mechanistic similarity to mammalian prions, they were termed yeast prions. Subsequent to this, a prion has also been found in the fungus Podospora anserina. These prions behave similarly to PrP, but, in general, are nontoxic to their hosts. Susan Lindquist's group at the Whitehead Institute has argued some of the fungal prions are not associated with any disease state, but may have a useful role; however, researchers at the NIH have also provided arguments suggesting that fungal prions could be considered a diseased state.[81] There is evidence that fungal proteins have evolved specific functions that are beneficial to the microorganism that enhance their ability to adapt to their diverse environments.[82]
Research into fungal prions has given strong support to the protein-only concept, since purified protein extracted from cells with a prion state has been demonstrated to convert the normal form of the protein into a misfolded form in vitro, and in the process, preserve the information corresponding to different strains of the prion state. It has also shed some light on prion domains, which are regions in a protein that promote the conversion into a prion. Fungal prions have helped to suggest mechanisms of conversion that may apply to all prions, though fungal prions appear distinct from infectious mammalian prions in the lack of cofactor required for propagation. The characteristic prion domains may vary between species – e.g., characteristic fungal prion domains are not found in mammalian prions.
| Protein | Natural host | Normal function | Prion state | Prion phenotype | Year identified |
|---|---|---|---|---|---|
| Ure2p | Saccharomyces cerevisiae | Nitrogen catabolite repressor | [URE3] | Growth on poor nitrogen sources | 1994 |
| Sup35p | S. cerevisiae | Translation termination factor | [PSI+] | Increased levels of nonsense suppression | 1994 |
| HET-S | Podospora anserina | Regulates heterokaryon incompatibility | [Het-s] | Heterokaryon formation between incompatible strains | |
| Rnq1p | S. cerevisiae | Protein template factor | [RNQ+], [PIN+] | Promotes aggregation of other prions | |
| Swi1 | S. cerevisiae | Chromatin remodeling | [SWI+] | Poor growth on some carbon sources | 2008 |
| Cyc8 | S. cerevisiae | Transcriptional repressor | [OCT+] | Transcriptional derepression of multiple genes | 2009 |
| Mot3 | S. cerevisiae | Nuclear transcription factor | [MOT3+] | Transcriptional derepression of anaerobic genes | 2009 |
| Sfp1 | S. cerevisiae | Putative transcription factor | [ISP+] | Antisuppression | 2010[83] |
6. Treatments
There are no effective treatments for prion diseases.[84] Clinical trials in humans have not met with success and have been hampered by the rarity of prion diseases.[84] Although some potential treatments have shown promise in the laboratory, none have been effective once the disease has commenced.[85]
7. In Other Diseases
Prion-like domains have been found in a variety of other mammalian proteins. Some of these proteins have been implicated in the ontogeny of age-related neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U), Alzheimer's disease, Parkinson's disease, and Huntington's disease.[86][87][88] They are also implicated in some forms of systemic amyloidosis including AA amyloidosis that develops in humans and animals with inflammatory and infectious diseases such as tuberculosis, Crohn's disease, rheumatoid arthritis, and HIV/AIDS. AA amyloidosis, like prion disease, may be transmissible.[89] This has given rise to the 'prion paradigm', where otherwise harmless proteins can be converted to a pathogenic form by a small number of misfolded, nucleating proteins.[90]
The definition of a prion-like domain arises from the study of fungal prions. In yeast, prionogenic proteins have a portable prion domain that is both necessary and sufficient for self-templating and protein aggregation. This has been shown by attaching the prion domain to a reporter protein, which then aggregates like a known prion. Similarly, removing the prion domain from a fungal prion protein inhibits prionogenesis. This modular view of prion behaviour has led to the hypothesis that similar prion domains are present in animal proteins, in addition to PrP.[86] These fungal prion domains have several characteristic sequence features. They are typically enriched in asparagine, glutamine, tyrosine and glycine residues, with an asparagine bias being particularly conducive to the aggregative property of prions. Historically, prionogenesis has been seen as independent of sequence and only dependent on relative residue content. However, this has been shown to be false, with the spacing of prolines and charged residues having been shown to be critical in amyloid formation.[91]
Bioinformatic screens have predicted that over 250 human proteins contain prion-like domains (PrLD). These domains are hypothesized to have the same transmissible, amyloidogenic properties of PrP and known fungal proteins. As in yeast, proteins involved in gene expression and RNA binding seem to be particularly enriched in PrLD's, compared to other classes of protein. In particular, 29 of the known 210 proteins with an RNA recognition motif also have a putative prion domain. Meanwhile, several of these RNA-binding proteins have been independently identified as pathogenic in cases of ALS, FTLD-U, Alzheimer's disease, and Huntington's disease.[92]
7.1. Role in Neurodegenerative Disease
The pathogenicity of prions and proteins with prion-like domains is hypothesized to arise from their self-templating ability and the resulting exponential growth of amyloid fibrils. The presence of amyloid fibrils in patients with degenerative diseases has been well documented. These amyloid fibrils are seen as the result of pathogenic proteins that self-propagate and form highly stable, non-functional aggregates.[92] While this does not necessarily imply a causal relationship between amyloid and degenerative diseases, the toxicity of certain amyloid forms and the overproduction of amyloid in familial cases of degenerative disorders supports the idea that amyloid formation is generally toxic.[93]
Specifically, aggregation of TDP-43, an RNA-binding protein, has been found in ALS/MND patients, and mutations in the genes coding for these proteins have been identified in familial cases of ALS/MND. These mutations promote the misfolding of the proteins into a prion-like conformation. The misfolded form of TDP-43 forms cytoplasmic inclusions in affected neurons, and is found depleted in the nucleus. In addition to ALS/MND and FTLD-U, TDP-43 pathology is a feature of many cases of Alzheimer's disease, Parkinson's disease and Huntington's disease. The misfolding of TDP-43 is largely directed by its prion-like domain. This domain is inherently prone to misfolding, while pathological mutations in TDP-43 have been found to increase this propensity to misfold, explaining the presence of these mutations in familial cases of ALS/MND. As in yeast, the prion-like domain of TDP-43 has been shown to be both necessary and sufficient for protein misfolding and aggregation.[86]
Similarly, pathogenic mutations have been identified in the prion-like domains of heterogeneous nuclear riboproteins hnRNPA2B1 and hnRNPA1 in familial cases of muscle, brain, bone and motor neuron degeneration. The wild-type form of all of these proteins show a tendency to self-assemble into amyloid fibrils, while the pathogenic mutations exacerbate this behaviour and lead to excess accumulation.[94]
8. Weaponization
Prions could theoretically be employed as a weaponized agent.[95][96] With potential fatality rates of 100%, prions could be an effective bio-weapon. An unfavorable aspect is prions' very long incubation periods. Persistent heavy exposure of prions to the intestine might shorten the overall onset.[97] Another aspect of using prions in warfare is the difficulty of detection and decontamination.[98]
9. History
In the 18th and 19th centuries, exportation of sheep from Spain was observed to coincide with a disease called scrapie. This disease caused the affected animals to "lie down, bite at their feet and legs, rub their backs against posts, fail to thrive, stop feeding and finally become lame".[99] The disease was also observed to have the long incubation period that is a key characteristic of transmissible spongiform encephalopathies (TSEs). Although the cause of scrapie was not known back then, it is probably the first transmissible spongiform encephalopathy to be recorded.
In the 1950s, Carleton Gajdusek began research which eventually showed that kuru could be transmitted to chimpanzees by what was possibly a new infectious agent, work for which he eventually won the 1976 Nobel prize. During the 1960s, two London-based researchers, radiation biologist Tikvah Alper and biophysicist John Stanley Griffith, developed the hypothesis that the transmissible spongiform encephalopathies are caused by an infectious agent consisting solely of proteins.[100][101] Earlier investigations by E.J. Field into scrapie and kuru had found evidence for the transfer of pathologically inert polysaccharides that only become infectious post-transfer, in the new host.[102][103] Alper and Griffith wanted to account for the discovery that the mysterious infectious agent causing the diseases scrapie and Creutzfeldt–Jakob disease resisted ionizing radiation.[104] Griffith proposed three ways in which a protein could be a pathogen.[105]
In the first hypothesis, he suggested that if the protein is the product of a normally suppressed gene, and introducing the protein could induce the gene's expression, that is, wake the dormant gene up, then the result would be a process indistinguishable from replication, as the gene's expression would produce the protein, which would then go wake the gene up in other cells.
His second hypothesis forms the basis of the modern prion theory, and proposed that an abnormal form of a cellular protein can convert normal proteins of the same type into its abnormal form, thus leading to replication. His third hypothesis proposed that the agent could be an antibody if the antibody was its own target antigen, as such an antibody would result in more and more antibody being produced against itself. However, Griffith acknowledged that this third hypothesis was unlikely to be true due to the lack of a detectable immune response.[106]
Francis Crick recognized the potential significance of the Griffith protein-only hypothesis for scrapie propagation in the second edition of his "Central dogma of molecular biology" (1970): While asserting that the flow of sequence information from protein to protein, or from protein to RNA and DNA was "precluded", he noted that Griffith's hypothesis was a potential contradiction (although it was not so promoted by Griffith).[107] The revised hypothesis was later formulated, in part, to accommodate reverse transcription (which both Howard Temin and David Baltimore discovered in 1970).[108]
In 1982, Stanley B. Prusiner of the University of California, San Francisco, announced that his team had purified the hypothetical infectious protein, which did not appear to be present in healthy hosts, though they did not manage to isolate the protein until two years after Prusiner's announcement.[9][109] The protein was named a prion, for "proteinacious infectious particle", derived from the words protein and infection. When the prion was discovered, Griffith's first hypothesis, that the protein was the product of a normally silent gene was favored by many. It was subsequently discovered, however, that the same protein exists in normal hosts but in different form.[110]
Following the discovery of the same protein in different form in uninfected individuals, the specific protein that the prion was composed of was named the prion protein (PrP), and Griffith's second hypothesis that an abnormal form of a host protein can convert other proteins of the same type into its abnormal form, became the dominant theory.[106] Prusiner won the Nobel Prize in Physiology or Medicine in 1997 for his research into prions.[111][112]
The content is sourced from: https://handwiki.org/wiki/Medicine:Prion
References
- "Prion". Merriam-Webster Dictionary. https://www.merriam-webster.com/dictionary/prion.
- "prion". Dictionary.com Unabridged. Random House. https://www.dictionary.com/browse/prion.
- "Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism". Proceedings of the National Academy of Sciences of the United States of America 112 (38): E5308–17. September 2015. doi:10.1073/pnas.1514475112. PMID 26324905. Bibcode: 2015PNAS..112E5308P. Lay summary: Makin, Simon (September 1, 2015). "A Red Flag for a Neurodegenerative Disease That May Be Transmissible". http://www.scientificamerican.com/article/a-red-flag-for-a-neurodegenerative-disease-that-may-be-transmissible/.
- "Stanley B. Prusiner – Autobiography". NobelPrize.org. http://nobelprize.org/nobel_prizes/medicine/laureates/1997/prusiner-autobio.html.
- "Etymologia: prion". Emerging Infectious Diseases 18 (6): 1030–1031. June 2012. doi:10.3201/eid1806.120271. PMID 22607731. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3381685
- "Dorland's Illustrated Medical Dictionary". Elsevier. http://dorlands.com/.
- "Merriam-Webster's Unabridged Dictionary". Merriam-Webster. http://unabridged.merriam-webster.com/unabridged/.
- Houghton Mifflin Harcourt. "The American Heritage Dictionary of the English Language". Houghton Mifflin Harcourt. https://ahdictionary.com/.
- "Novel proteinaceous infectious particles cause scrapie". Science 216 (4542): 136–44. April 1982. doi:10.1126/science.6801762. PMID 6801762. Bibcode: 1982Sci...216..136P. https://pdfs.semanticscholar.org/f292/b22e2675419c6392a5e55f6b35b1dfc46917.pdf.
- "Biomedicine. A view from the top – prion diseases from 10,000 feet". Science 300 (5621): 917–919. May 2003. doi:10.1126/science.1085920. PMID 12738843. https://zenodo.org/record/1230830. Retrieved 2020-07-28.
- "Cellular prion protein is released on exosomes from activated platelets". Blood 107 (10): 3907–3911. 15 May 2006. doi:10.1182/blood-2005-02-0802. PMID 16434486. https://dx.doi.org/10.1182%2Fblood-2005-02-0802
- "A transmembrane form of the prion protein in neurodegenerative disease". Science 279 (5352): 827–34. February 1998. doi:10.1126/science.279.5352.827. PMID 9452375. Bibcode: 1998Sci...279..827H. http://pdfs.semanticscholar.org/4320/9efc152784dbc7f0b9a1300d0ec9be602a2c.pdf.
- "Taking aim at the transmissible spongiform encephalopathie's infectious agents". Prions and mad cow disease. New York: Marcel Dekker. 2004. p. 6. ISBN 978-0-8247-4083-2. https://books.google.com/books?id=WjeuaHopV5UC&pg=PA6. Retrieved 2020-06-02.
- "The cellular prion protein binds copper in vivo". Nature 390 (6661): 684–87. 1997. doi:10.1038/37783. PMID 9414160. Bibcode: 1997Natur.390..684B. https://dx.doi.org/10.1038%2F37783
- "The state of the prion". Nature Reviews. Microbiology 2 (11): 861–71. November 2004. doi:10.1038/nrmicro1025. PMID 15494743. https://dx.doi.org/10.1038%2Fnrmicro1025
- "Regulation of embryonic cell adhesion by the prion protein". PLOS Biology 7 (3): e55. March 2009. doi:10.1371/journal.pbio.1000055. PMID 19278297. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2653553
- "Biochemistry and structure of PrP(C) and PrP(Sc)". British Medical Bulletin 66 (1): 21–33. 2003-06-01. doi:10.1093/bmb/66.1.21. PMID 14522846. https://dx.doi.org/10.1093%2Fbmb%2F66.1.21
- "Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding". Nature 411 (6839): 810–3. June 2001. doi:10.1038/35081095. PMID 11459061. Bibcode: 2001Natur.411..810S. https://dx.doi.org/10.1038%2F35081095
- "Autocatalytic self-propagation of misfolded prion protein". Proceedings of the National Academy of Sciences of the United States of America 101 (33): 12207–11. August 2004. doi:10.1073/pnas.0404650101. PMID 15297610. Bibcode: 2004PNAS..10112207B. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=514458
- "Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins". Proceedings of the National Academy of Sciences of the United States of America 90 (23): 10962–66. December 1993. doi:10.1073/pnas.90.23.10962. PMID 7902575. Bibcode: 1993PNAS...9010962P. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=47901
- "Cross-species transmission of CWD prions". Prion 10 (1): 83–91. 2016. doi:10.1080/19336896.2015.1118603. PMID 26809254. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4981193
- "Healthy prions protect nerves". Nature. 2010-01-24. doi:10.1038/news.2010.29. https://dx.doi.org/10.1038%2Fnews.2010.29
- "Necroptosis in anti-viral inflammation". Nature 26 (1): 4–13. 2019. doi:10.1038/s41418-018-0172-x. PMID 30050058. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6294789
- "Prions as adaptive conduits of memory and inheritance". Nature Reviews Genetics 6 (6): 435–50. June 2005. doi:10.1038/nrg1616. PMID 15931169. https://dx.doi.org/10.1038%2Fnrg1616
- "Hippocampal synaptic plasticity in mice devoid of cellular prion protein". Brain Research. Molecular Brain Research 131 (1–2): 58–64. November 2004. doi:10.1016/j.molbrainres.2004.08.004. PMID 15530652. https://dx.doi.org/10.1016%2Fj.molbrainres.2004.08.004
- "PrPC controls via protein kinase A the direction of synaptic plasticity in the immature hippocampus". The Journal of Neuroscience 33 (7): 2973–83. February 2013. doi:10.1523/JNEUROSCI.4149-12.2013. PMID 23407955. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6619229
- "Long-term memory consolidation: The role of RNA-binding proteins with prion-like domains". RNA Biology 14 (5): 568–86. 2016-10-11. doi:10.1080/15476286.2016.1244588. PMID 27726526. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5449092
- "Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal". Proceedings of the National Academy of Sciences of the United States of America 103 (7): 2184–89. February 2006. doi:10.1073/pnas.0510577103. PMID 16467153. Bibcode: 2006PNAS..103.2184Z. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1413720
- "Prion Protein PRNP: A New Player in Innate Immunity? The Aβ Connection". Journal of Alzheimer's Disease Reports 1 (1): 263–275. December 2017. doi:10.3233/ADR-170037. PMID 30480243. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6159716
- "Structural clues to prion replication". Science 264 (5158): 530–31. April 1994. doi:10.1126/science.7909169. PMID 7909169. Bibcode: 1994Sci...264..530C. https://dx.doi.org/10.1126%2Fscience.7909169
- "Prionics or the kinetic basis of prion diseases". Biophysical Chemistry 63 (1): A1–18. December 1996. doi:10.1016/S0301-4622(96)02250-8. PMID 8981746. https://dx.doi.org/10.1016%2FS0301-4622%2896%2902250-8
- "Copurification of Sp33-37 and scrapie agent from hamster brain prior to detectable histopathology and clinical disease". The Journal of General Virology 72 (12): 2905–13. December 1991. doi:10.1099/0022-1317-72-12-2905. PMID 1684986. https://dx.doi.org/10.1099%2F0022-1317-72-12-2905
- "Proteinase-resistant prion protein accumulation in Syrian hamster brain correlates with regional pathology and scrapie infectivity". Neurology 41 (9): 1482–90. September 1991. doi:10.1212/WNL.41.9.1482. PMID 1679911. https://dx.doi.org/10.1212%2FWNL.41.9.1482
- "Sequential appearance and accumulation of pathognomonic markers in the central nervous system of hamsters orally infected with scrapie". The Journal of General Virology 77 (8): 1925–34. August 1996. doi:10.1099/0022-1317-77-8-1925. PMID 8760444. https://dx.doi.org/10.1099%2F0022-1317-77-8-1925
- "Prion protein structure and scrapie replication: theoretical, spectroscopic, and genetic investigations". Cold Spring Harbor Symposia on Quantitative Biology 61: 495–509. 1996. doi:10.1101/SQB.1996.061.01.050. PMID 9246476. https://dx.doi.org/10.1101%2FSQB.1996.061.01.050
- "Quantifying the kinetic parameters of prion replication". Biophysical Chemistry 77 (2–3): 139–52. March 1999. doi:10.1016/S0301-4622(99)00016-2. PMID 10326247. https://dx.doi.org/10.1016%2FS0301-4622%2899%2900016-2
- "An analytical solution to the kinetics of breakable filament assembly". Science 326 (5959): 1533–37. December 2009. doi:10.1126/science.1178250. PMID 20007899. Bibcode: 2009Sci...326.1533K. https://dx.doi.org/10.1126%2Fscience.1178250
- "Designing drugs to stop the formation of prion aggregates and other amyloids". Biophysical Chemistry 88 (1–3): 47–59. December 2000. doi:10.1016/S0301-4622(00)00197-6. PMID 11152275. https://dx.doi.org/10.1016%2FS0301-4622%2800%2900197-6
- "Formation of native prions from minimal components in vitro". Proceedings of the National Academy of Sciences of the United States of America 104 (23): 9741–6. June 2007. doi:10.1073/pnas.0702662104. PMID 17535913. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1887554
- "Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions". Proceedings of the National Academy of Sciences of the United States of America 109 (28): E1938-46. July 2012. doi:10.1073/pnas.1206999109. PMID 22711839. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3396481
- "90. Prions". ICTVdB Index of Viruses. U.S. National Institutes of Health website. 2002-02-14. https://www.ncbi.nlm.nih.gov/ICTVdb/Ictv/fs_prion.htm.
- "Prion Disease in Dromedary Camels, Algeria". Emerging Infectious Diseases 24 (6): 1029–36. June 2018. doi:10.3201/eid2406.172007. PMID 29652245. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6004840
- "Prion Diseases: A Review; II. Prion Diseases in Man and Animals". Scientific Journal of King Faisal University (Basic and Applied Sciences) 5 (2): 139. 2004. https://apps.kfu.edu.sa/sjournal/ara/pdffiles/b526.pdf. Retrieved April 9, 2016.
- "Prion protein conformation in a patient with sporadic fatal insomnia". The New England Journal of Medicine 340 (21): 1630–1638. May 1999. doi:10.1056/NEJM199905273402104. PMID 10341275. Lay summary: "BSE proteins may cause fatal insomnia". May 28, 1999. http://news.bbc.co.uk/2/hi/health/355297.stm.
- "Familial spongiform encephalopathy associated with a novel prion protein gene mutation". Annals of Neurology 42 (2): 138–46. August 1997. doi:10.1002/ana.410420203. PMID 9266722. https://dx.doi.org/10.1002%2Fana.410420203
- Robbins pathologic basis of disease. Philadelphia: Saunders. 1999. ISBN 072167335X.
- "Transmissible spongiform encephalopathies in humans". Annual Review of Microbiology 53: 283–314. 1999. doi:10.1146/annurev.micro.53.1.283. PMID 10547693. https://zenodo.org/record/1235027. Retrieved 2019-07-10.
- "Prion Diseases". US Centers for Disease Control. 2006-01-26. https://www.cdc.gov/ncidod/dvrd/prions/.
- "Prion diseases of humans and animals: their causes and molecular basis". Annual Review of Neuroscience 24: 519–50. 2001. doi:10.1146/annurev.neuro.24.1.519. PMID 11283320. http://pdfs.semanticscholar.org/650f/8f4c880880d357e5dd82236ba611065e21cc.pdf.
- "Variant Creutzfeldt–Jakob disease: risk of transmission by blood transfusion and blood therapies". Haemophilia 12 (Suppl 1): 8–15, discussion 26–28. March 2006. doi:10.1111/j.1365-2516.2006.01195.x. PMID 16445812. https://dx.doi.org/10.1111%2Fj.1365-2516.2006.01195.x
- "Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease". The EMBO Journal 20 (15): 3957–3966. August 2001. doi:10.1093/emboj/20.15.3957. PMID 11483499. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=149175
- "Mucosal vaccination delays or prevents prion infection via an oral route". Neuroscience 133 (2): 413–421. 2005. doi:10.1016/j.neuroscience.2005.02.031. PMID 15878645. Lay summary: "Active Vaccine Prevents Mice From Developing Prion Disease". May 14, 2005. https://www.sciencedaily.com/releases/2005/05/050514111648.htm.
- "Scientists Announce Mad Cow Breakthrough". The Washington Post. 2007-01-01. https://www.washingtonpost.com/wp-dyn/content/article/2006/12/31/AR2006123100672.html. "Scientists said yesterday that they have used genetic engineering techniques to produce the first cattle that may be biologically incapable of getting mad cow disease"
- "Mice devoid of PrP are resistant to scrapie". Cell 73 (7): 1339–1347. July 1993. doi:10.1016/0092-8674(93)90360-3. PMID 8100741. https://dx.doi.org/10.1016%2F0092-8674%2893%2990360-3
- "Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey". BMJ 347: f5675. October 2013. doi:10.1136/bmj.f5675. PMID 24129059. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3805509
- "Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers". Nature 457 (7233): 1128–32. February 2009. doi:10.1038/nature07761. PMID 19242475. Bibcode: 2009Natur.457.1128L. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2748841
- "Protein Misfolding Cyclic Amplification of Infectious Prions". Progress in Molecular Biology and Translational Science 150: 361–374. 2017. doi:10.1016/bs.pmbts.2017.06.016. ISBN 9780128112267. PMID 28838669. https://dx.doi.org/10.1016%2Fbs.pmbts.2017.06.016
- Prion Diseases Diagnosis and Pathogeneis. Archives of Virology. 16. New York: Springer. 2001. ISBN 978-3211835302.
- "Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein". Cell 83 (1): 79–90. October 1995. doi:10.1016/0092-8674(95)90236-8. PMID 7553876. https://dx.doi.org/10.1016%2F0092-8674%2895%2990236-8
- "Oral transmissibility of prion disease is enhanced by binding to soil particles". PLOS Pathogens 3 (7): e93. July 2007. doi:10.1371/journal.ppat.0030093. PMID 17616973. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1904474
- "Asymptomatic deer excrete infectious prions in faeces". Nature 461 (7263): 529–532. September 2009. doi:10.1038/nature08289. PMID 19741608. Bibcode: 2009Natur.461..529T. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3186440
- "Aerosols transmit prions to immunocompetent and immunodeficient mice". PLOS Pathogens 7 (1): e1001257. January 2011. doi:10.1371/journal.ppat.1001257. PMID 21249178. Lay summary: Mackenzie, Deborah (January 13, 2011). "Prion disease can spread through air". https://www.newscientist.com/article/dn19971-prion-disease-can-spread-through-air.
- "Detection of prion protein in urine-derived injectable fertility products by a targeted proteomic approach". PLOS ONE 6 (3): e17815. March 2011. doi:10.1371/journal.pone.0017815. PMID 21448279. Bibcode: 2011PLoSO...617815V. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3063168
- "Surprising' Discovery Made About Chronic Wasting Disease". Food Safety News. June 1, 2015. http://www.foodsafetynews.com/2015/06/researchers-make-surprising-discovery-about-spread-of-chronic-wasting-disease/.
- "Grass plants bind, retain, uptake, and transport infectious prions". Cell Reports 11 (8): 1168–75. May 2015. doi:10.1016/j.celrep.2015.04.036. PMID 25981035. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4449294
- "Doppel: more rival than double to prion". Neuroscience 141 (1): 1–8. August 2006. doi:10.1016/j.neuroscience.2006.04.057. PMID 16781817. https://dx.doi.org/10.1016%2Fj.neuroscience.2006.04.057
- "Inactivation of transmissible spongiform encephalopathy (prion) agents by environ LpH". Journal of Virology 78 (4): 2164–65. February 2004. doi:10.1128/JVI.78.4.2164-2165.2004. PMID 14747583. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=369477
- "Methods to minimize the risks of Creutzfeldt–Jakob disease transmission by surgical procedures: where to set the standard?". Clinical Infectious Diseases 43 (6): 757–64. September 2006. doi:10.1086/507030. PMID 16912952. https://dx.doi.org/10.1086%2F507030
- "Transmissible spongiform encephalopathies". Lancet 363 (9402): 51–61. January 2004. doi:10.1016/S0140-6736(03)15171-9. PMID 14723996. https://dx.doi.org/10.1016%2FS0140-6736%2803%2915171-9
- "New studies on the heat resistance of hamster-adapted scrapie agent: threshold survival after ashing at 600 degrees C suggests an inorganic template of replication". Proceedings of the National Academy of Sciences of the United States of America 97 (7): 3418–21. March 2000. doi:10.1073/pnas.050566797. PMID 10716712. Bibcode: 2000PNAS...97.3418B. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=16254
- "Ozone Sterilization". UK Health Protection Agency. 2005-04-14. http://www.hpa.org.uk/hpa/news/articles/press_releases/2005/050414_ozone_sterilizer.htm.
- "Rapid chemical decontamination of infectious CJD and scrapie particles parallels treatments known to disrupt microbes and biofilms". Virulence 6 (8): 787–801. 2015. doi:10.1080/21505594.2015.1098804. PMID 26556670. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4826107
- "Proteolysis of abnormal prion protein with a thermostable protease from Thermococcus kodakarensis KOD1". Applied Microbiology and Biotechnology 98 (5): 2113–2120. March 2014. doi:10.1007/s00253-013-5091-7. PMID 23880875. https://dx.doi.org/10.1007%2Fs00253-013-5091-7
- "A Novel, Reliable and Highly Versatile Method to Evaluate Different Prion Decontamination Procedures". Frontiers in Bioengineering and Biotechnology 8: 589182. 2020. doi:10.3389/fbioe.2020.589182. PMID 33195153. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=7658626
- "Transmission of prions". Proceedings of the National Academy of Sciences of the United States of America 99 (Suppl 4): 16378–83. December 2002. doi:10.1073/pnas.172403799. PMID 12181490. Bibcode: 2002PNAS...9916378W. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=139897
- "The Ecology of Prions". Microbiology and Molecular Biology Reviews 81 (3). September 2017. doi:10.1128/MMBR.00001-17. PMID 28566466. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5584314
- "Soil humic acids degrade CWD prions and reduce infectivity". PLOS Pathogens 14 (11): e1007414. November 2018. doi:10.1371/journal.ppat.1007414. PMID 30496301. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6264147
- "Mitigation of prion infectivity and conversion capacity by a simulated natural process--repeated cycles of drying and wetting". PLOS Pathogens 11 (2): e1004638. February 2015. doi:10.1371/journal.ppat.1004638. PMID 25665187. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4335458
- "Investigating protein conformation-based inheritance and disease in yeast". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 356 (1406): 169–76. February 2001. doi:10.1098/rstb.2000.0762. PMID 11260797. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1088422
- "Unraveling prion strains with cell biology and organic chemistry". Proceedings of the National Academy of Sciences of the United States of America 105 (1): 11–12. January 2008. doi:10.1073/pnas.0710824105. PMID 18172195. Bibcode: 2008PNAS..105...11A. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2224168
- "Probing the role of PrP repeats in conformational conversion and amyloid assembly of chimeric yeast prions". The Journal of Biological Chemistry 282 (47): 34204–12. November 2007. doi:10.1074/jbc.M704952200. PMID 17893150. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2262835
- "Blessings in disguise: biological benefits of prion-like mechanisms". Trends in Cell Biology 23 (6): 251–59. June 2013. doi:10.1016/j.tcb.2013.01.007. PMID 23485338. https://dx.doi.org/10.1016%2Fj.tcb.2013.01.007
- "Non-Mendelian determinant [ISP+ in yeast is a nuclear-residing prion form of the global transcriptional regulator Sfp1"]. Proceedings of the National Academy of Sciences of the United States of America 107 (23): 10573–77. June 2010. doi:10.1073/pnas.1005949107. PMID 20498075. Bibcode: 2010PNAS..10710573R. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2890785
- "Toward Therapy of Human Prion Diseases". Annual Review of Pharmacology and Toxicology 58: 331–51. January 2018. doi:10.1146/annurev-pharmtox-010617-052745. PMID 28961066. https://www.zora.uzh.ch/id/eprint/141186/1/Aguzzi_et_al%3B_2017_revised.pdf. Retrieved 2020-03-05.
- "Prion Clinic – Drug treatments". 13 September 2017. http://www.prion.ucl.ac.uk/clinic-services/research/drug-treatments/.
- "The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease". Brain Research 1462: 61–80. June 2012. doi:10.1016/j.brainres.2012.01.016. PMID 22445064. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3372647
- "NEURODEGENERATION. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein". Science 349 (6248): 1255555. August 2015. doi:10.1126/science.1255555. PMID 26250687. https://dx.doi.org/10.1126%2Fscience.1255555
- "Parkinson's disease and alpha synuclein: is Parkinson's disease a prion-like disorder?". Movement Disorders 28 (1): 31–40. January 2013. doi:10.1002/mds.25373. PMID 23390095. https://dx.doi.org/10.1002%2Fmds.25373
- "Transmission of systemic AA amyloidosis in animals". Veterinary Pathology 51 (2): 363–71. March 2014. doi:10.1177/0300985813511128. PMID 24280941. https://dx.doi.org/10.1177%2F0300985813511128
- "Self-propagation of pathogenic protein aggregates in neurodegenerative diseases". Nature 501 (7465): 45–51. September 2013. doi:10.1038/nature12481. PMID 24005412. Bibcode: 2013Natur.501...45J. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3963807
- "A systematic survey identifies prions and illuminates sequence features of prionogenic proteins". Cell 137 (1): 146–58. 2009. doi:10.1016/j.cell.2009.02.044. PMID 19345193. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2683788
- "The amyloid state of proteins in human diseases". Cell 148 (6): 1188–203. March 2012. doi:10.1016/j.cell.2012.02.022. PMID 22424229. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3353745
- "Prion protein - mediator of toxicity in multiple proteinopathies". Nature Reviews. Neurology 16 (4): 187–188. April 2020. doi:10.1038/s41582-020-0332-8. PMID 32123368. https://dx.doi.org/10.1038%2Fs41582-020-0332-8
- "Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS". Nature 495 (7442): 467–73. March 2013. doi:10.1038/nature11922. PMID 23455423. Bibcode: 2013Natur.495..467K. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3756911
- "What are Biological Weapons?". United Nations, Office for Disarmament Affairs. https://www.un.org/disarmament/biological-weapons/about/what-are-biological-weapons/.
- "Prions: the danger of biochemical weapons". http://www.scielo.br/pdf/cta/v34n3/01.pdf.
- "The Next Plague: Prions are Tiny, Mysterious and Frightening". American Council on Science and Health. 20 March 2017. https://www.acsh.org/news/2017/03/20/next-plague-prions-are-tiny-mysterious-and-frightening-11018.
- "Prions as Bioweapons? - Much Ado About Nothing; or Apt Concerns Over Tiny Proteins used in Biowarfare". Defence iQ. 13 September 2019. https://www.defenceiq.com/air-land-and-sea-defence-services/articles/prions-as-bioweapons.
- "How Prions Came to Be: A Brief History – Infectious Disease: Superbugs, Science, & Society" (in en-US). https://sites.duke.edu/superbugs/module-6/prions-mad-cow-disease-when-proteins-go-bad/how-prions-came-to-be-a-brief-history/.
- "Does the agent of scrapie replicate without nucleic acid?". Nature 214 (5090): 764–66. May 1967. doi:10.1038/214764a0. PMID 4963878. Bibcode: 1967Natur.214..764A. https://dx.doi.org/10.1038%2F214764a0
- "Self-replication and scrapie". Nature 215 (5105): 1043–44. September 1967. doi:10.1038/2151043a0. PMID 4964084. Bibcode: 1967Natur.215.1043G. https://dx.doi.org/10.1038%2F2151043a0
- "Transmission experiments with multiple sclerosis: an interim report". British Medical Journal 2 (5513): 564–65. September 1966. doi:10.1136/bmj.2.5513.564. PMID 5950508. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1943767
- "The infective process in scrapie". Lancet 2 (7570): 714–16. September 1968. doi:10.1016/s0140-6736(68)90754-x. PMID 4175093. https://dx.doi.org/10.1016%2Fs0140-6736%2868%2990754-x
- "Susceptibility of scrapie agent to ionizing radiation". Nature. 5188 222 (5188): 90–91. April 1969. doi:10.1038/222090a0. PMID 4975649. Bibcode: 1969Natur.222...90F. https://dx.doi.org/10.1038%2F222090a0
- "Self-replication and scrapie". Nature 215 (5105): 1043–44. Sep 1967. doi:10.1038/2151043a0. PMID 4964084. Bibcode: 1967Natur.215.1043G. https://dx.doi.org/10.1038%2F2151043a0
- "Prions, the Protein Hypothesis, and Scientific Revolutions". Prions and Mad Cow Disease. Marcel Dekker. January 1, 2004. pp. 21–60. ISBN 978-0-203-91297-3. https://www.researchgate.net/publication/235220355. Retrieved July 27, 2018.
- "Central dogma of molecular biology". Nature 227 (5258): 561–63. August 1970. doi:10.1038/227561a0. PMID 4913914. Bibcode: 1970Natur.227..561C. https://dx.doi.org/10.1038%2F227561a0
- "The Discovery of Reverse Transcriptase". Annual Review of Virology 3 (1): 29–51. September 2016. doi:10.1146/annurev-virology-110615-035556. PMID 27482900. https://dx.doi.org/10.1146%2Fannurev-virology-110615-035556
- "The game of name is fame. But is it science?". Discover 7 (12): 28–41. December 1986.
- "Prion protein scrapie and the normal cellular prion protein". Prion 10 (1): 63–82. 2016. doi:10.1080/19336896.2015.1110293. PMID 26645475. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4981215
- "The Nobel Prize in Physiology or Medicine, 1997". NobelPrize.org. http://nobelprize.org/nobel_prizes/medicine/laureates/1997/. "The Nobel Prize in Physiology or Medicine 1997 was awarded to Stanley B. Prusiner 'for his discovery of Prions - a new biological principle of infection.'"
- "Prions Are Forever" (in en). https://blogs.scientificamerican.com/artful-amoeba/prions-are-forever/.