Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Ergot is the spore form of the fungus Claviceps purpurea. Ergot alkaloids are indole compounds that are biosynthetically derived from L-tryptophan and represent the largest group of fungal nitrogen metabolites found in nature. The common part of ergot alkaloids is lysergic acid.
- lysergic acid
- ergot alkaloids
- total synthesis
1. Lysergic Acid as a Component of Ergot Alkaloids
1.1. Ergot
Ergot is the spore form of the fungus Claviceps purpurea (Fr.) Tul. (see Section Abbreviations used in the text). It occurs in the form of elongated sclerotia, ranging in length from a few millimeters to 4–6 cm. Ergot, in addition to causing diseases of cereal and grass, and, hence, lowering yields, also causes grain contamination with toxic substances, alkaloids, which are harmful to both humans and animals, causing a number of diseases commonly referred to as “ergotism” [1].
1.1.1. History of Ergot [2]
The problem of the presence of ergot and its toxicity has existed since the agrarian revolution. There is historical evidence of their presence in numerous cultures, ranging from the Assyrians (600 BC), through ancient Rome (1st century BC), to medieval Europe. Typically, ergot has been described as “harmful spots on the spikes” and “poisonous grass causing miscarriage”. Hippocrates identified a plant disease in the 4th century BC, which he called melanthion—and described the inhibitory effect of ergot on postpartum bleeding [3].
Ergotic epidemics frequently occurred in medieval Europe. They were due to the prevalence of contaminated rye used in baking bread. Rye often contained large amounts of alkaloids that induced ergotism. The very first ergot epidemic was mentioned in Annales Xantensen (Germany, 857). Numerous sources describe successive epidemics in France, Germany, and Scandinavia. The 16th century was particularly dramatic, when tens of thousands of people died in Europe from ergot poisoning. The last major epidemic took place in Russia in 1926–1927 in the Ural region. Single cases of ergotamine poisoning have occurred since then [1][4].
One of the first to make connection between illness and eating contaminated grain was the German physician Wendelin Thelius, in 1596. In 1670, Thuillier linked the presence of ergot in rye bread with epidemics by experimenting with feeding ergot to animals, which led to their death. At the end of the 18th century, L’Abbe Tessier proposed preventive measures—compulsory grain cleaning and replacing contaminated grain with potatoes in the diet. It was not until 1764 that von Münchhausen stated that ergot was a fungus [1].
Ergot was also used for medicinal purposes. Ergot alkaloids were used as early as 2000 BC in Greece as a hallucinatory agent, in Mesopotamia as an ingredient of “magic spells”, and in Egypt in a formulation described as a hair growth remedy. The first confirmed reports of the deliberate use of ergot come from around 1100 B.C.E. from China, where it was used in obstetrics and gynecology. In the 16th century, ergot was also used for medical purposes in the West—Adam Lonicer used it to induce labor, and Thalius used ergot to stanch the bleeding. In 1820, ergot was introduced into the United States Pharmacopoeia [1].
Nowadays, the presence of ergot in cereal is no longer a problem. Grain cleaning techniques used in mills remove up to 82% of the ergot contained in them. In addition, the process of baking wheat bread allows for the reduction of the concentration of alkaloids, even to zero [4].
1.1.2. Properties of Ergot
As described above, ergot is both toxic and therapeutic [4]. Two different forms of ergot poisoning have been described: ergotismus gangrenosus (gangrene, “St. Anthony’s fire”) and ergotismus convulsivus (contractile form, “St. Vitus dance”). The two forms can coexist or be independent of each other. Symptoms present in gangrenous form, include diarrhea, sensory disturbances, slow heart rate, shortness of breath, paralysis of the respiratory center, ischemia affecting the limbs, thrombotic changes followed by gangrene, loss of limbs, and death. In the case of the systolic form, symptoms comprise of sensory disturbances, muscle tremors, headache, abdominal pain, slow heart rate, and, at the same time, strong nervous excitation [3][4].
Ergot is also toxic to animals, causing symptoms similar to those observed in humans: loss of milk, convulsive ergotism, and gangrene.
The therapeutic effects of ergot may include inhibition of prolactin, treatment of parkinsonism [5], treatment of cerebrovascular insufficiency, migraine, venous insufficiency, thrombosis, embolism, stimulation of cerebral, peripheral metabolism, and stimulation of the uterus [1][4].
Moreover, ergot alkaloids have been used to induce abortion for hundreds of years [3].
1.2. Ergot Alkaloids
1.2.1. Types of Ergot Alkaloids
Ergot alkaloids are based on the ergoline ring system (Figure 1).
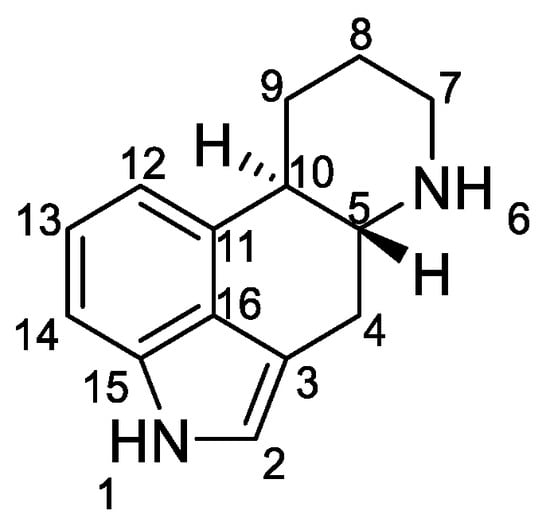
Figure 1. Structure of ergoline.
Ergoline derivatives have two asymmetric centers, either at the C-5 and C-10 positions or at the C-5 and C-8 positions. The stereochemistry and configuration of all atoms in the structures are accurately described in the literature, in particular the relative positions of the hydrogen [6][7].
Ergot alkaloids are indole compounds that are biosynthetically derived from L-tryptophan and represents the largest group of fungal nitrogen metabolites found in nature. Ergot contains 0.15% to 0.5% of alkaloids, which can be separated by analytical methods into individual ones, with more than 80 individual ergot alkaloids [2].
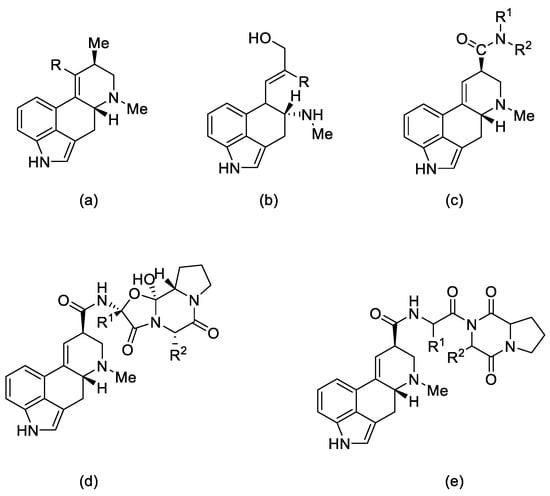
Figure 2. Structural formulas of the 5 main groups of ergot alkaloids: (a) clavine alkaloids and (b) 6,7-secoergolenes (alkaloids with an open D ring), (c) lysergic acid derivatives, (d) peptide alkaloids, and (e) lactam ergot alkaloids.
Ergot alkaloids can also be classified according to their molecular weight. Low molecular weight ergot alkaloids are amides of lysergic (or isolysergic) acid and amino alcohols. Ergometrine (ergobasine, ergonovine, ergoklinine, ergostetrine, and ergotocine) is an amide of 2-aminopropanol and lysergic acid, and ergometrinine (ergobasinine and isoergometrine) is an amide of isolysergic acid. High molecular weight ergot alkaloids are oligopeptides consisting of a ring of three amino acids (always with proline), linked to lysergic acid by a peptide bond. These are compounds such as: ergotamine, ergotin, ergosine, ergotoxin, ergocryptine, ergocristine, ergoclavine, and chanoclavine [1].
1.2.2. Biochemical and Pharmacological Properties
Ergot alkaloids—derivatives of D-lysergic acid—act by antagonistic or agonistic affinity to α adrenergic receptors as well as to serotonin receptors and dopamine receptors (D1–D5) [10]. This is due to the structural similarity to endogenous amines—noradrenaline, dopamine [11], and serotonin [12] (Figure 3).
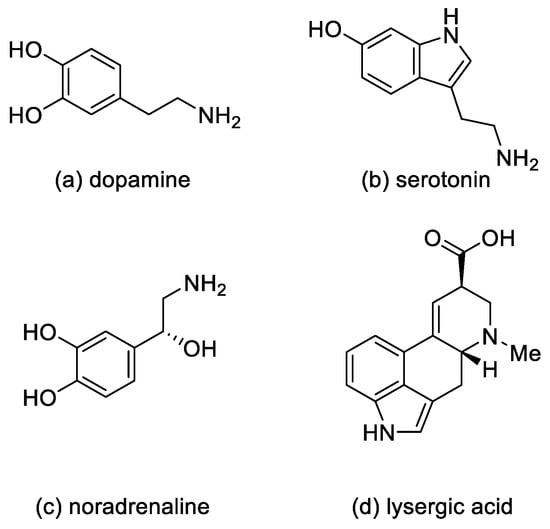
Figure 3. Structural formulas of endogenous amines and lysergic acid.
In medicine, ergotamine, dihydroergotamine, dihydroergotoxin, and dihydroergocristine, as well as their mixtures, are used [10].
These compounds are applied in medicine as sedatives and painkillers. They are also used as antispasmodics (in migraine attacks and in gynecology, in particular ergotamine); as drugs in orthostatic hypotension, in the treatment of intellectual disorders, and after strokes (for example, dihydroergotoxins) [10].
Lysergic acid diethylamide (LSD) (Figure 4) is a semisynthetic hallucinogenic compound. It is one of the most active psychedelic substances with hallucinogenic effects—the mild effects appear at a dose of 25 µg. LSD is an agonist of the 5-HT2A receptor located on the inhibitory interneurons of the brain. However, the action of LSD is not limited to that of the serotonin receptors. Although LSD and the other psychoactive derivatives exert their effects through activation of 5-HT2A receptors, they can have a significant behavioral component mediated by activation of 5-HT1A receptors [13][14]. Moreover, LSD also has an effect on the dopamine D1 and D2 receptors [15].
Figure 4. Ergot alkaloid derivatives.
Due to a short-term reduction in serotonin levels, after the LSD interaction with cells subsides, its production in the cells increases abruptly, which may cause disintegration of cortical–cortical neuronal activity—and, consequently, induce increased perception of sensory stimuli and cognitive impairment [16]. Interestingly, LSD is not addictive, and despite the fact that it is not currently used in medicine, there are ongoing studies on its use in psychiatry—e.g., in the fight against alcoholism [17][18].
1.2.3. Other Application [19]
According to the latest reports, a semisynthetic lysergic acid derivative containing a urethane bond at the C-8 substituent—metergoline (Figure 5), has an interesting antibacterial application.
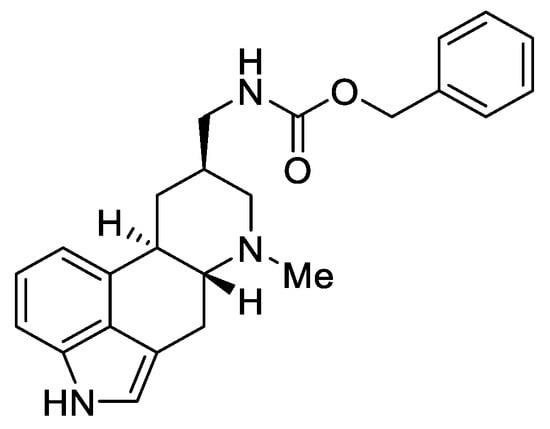
Figure 5. Metergoline.
This compound is authorized under the trade names Contralac and Liserdol as a hyperprolactinemia drug [20]. Previous studies have indicated its agonistic activity towards the 5 HT1A, 5-HT1D, and 5-HT1B receptors, as well as antagonistic activity towards the 5-HT2A, 5-HT2B, 5-HT2C, and 5-HT7 receptors [21][22].
The latest data, published in 2022 by Brown and Magolan [19], indicate that metergoline is an inhibitor of the intracellular Gram-negative pathogen Salmonella typhimurium. The biomechanism of action assumes a change in the permeability of the outer membrane of SPR741, which allows for an increase in the potential of biological activity against the MRSA strains, WT E. coli, Acinetobacter baumannii, and Burkholderia cenocepacia. The authors suggest that the ergot scaffold (especially the amide derivatives of lysergic acid, such as metergoline) may be the basis for the production of new efficient antibiotics.
1.3. Lysergic Acid
Lysergic acid (Figure 6) is a chemical compound based on the structure of ergoline—a four-ring system—which has a COOH acid group in the C-8 position and a methylamine in the 6-position in the D ring.
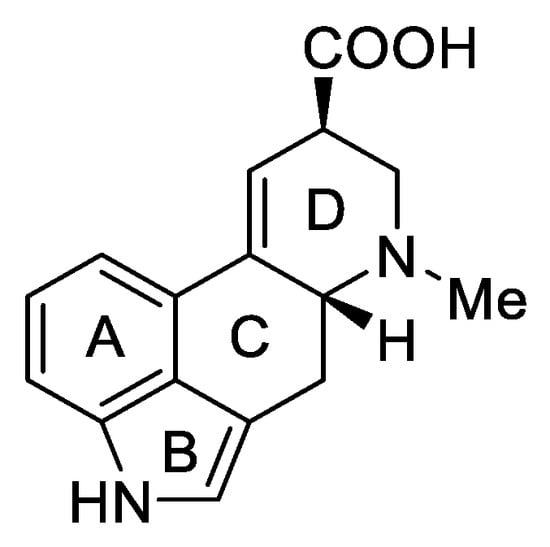
Figure 6. d-Lysergic acid.
Lysergic acid is a very important chemical compound obtained on a large scale. This is because it is a key precursor for the preparation of more complex ergot alkaloids, especially lysergic acid amides.
History of Lysergic Acid [23]
The isolation and analysis of ergot alkaloids began in 1906, when ergotoxine (a mixture of three proline-based alkaloids, ergocristine, ergocriptine, and ergocornine) was isolated from ergot [24], and its adrenolytic activity was discovered [25]. Twelve years later, Stoll isolated ergotamine—the first chemically pure ergot alkaloid, with major medical uses [26]. From that moment, the research efforts intensified. Since 1934, comprehensive studies of ergot alkaloids have been carried out by way of collaboration between teams in the USA, England, and Switzerland, with pharmaceutical companies investigating the pharmacological and clinical effects [27]. As a result, Jacobs and Craig identified the common part for ergot alkaloids—lysergic acid [28].
Lysergic acid is a chiral compound with two stereogenic centers. Subsequently, it was possible to identify all four stereoisomeric forms in which this acid can occur: d-lysergic acid, l-lysergic acid, d-isolysergic acid, and l-isolysergic acid; this has been described by Stoll, Hofmann, and Troxler (Figure 7) [29].
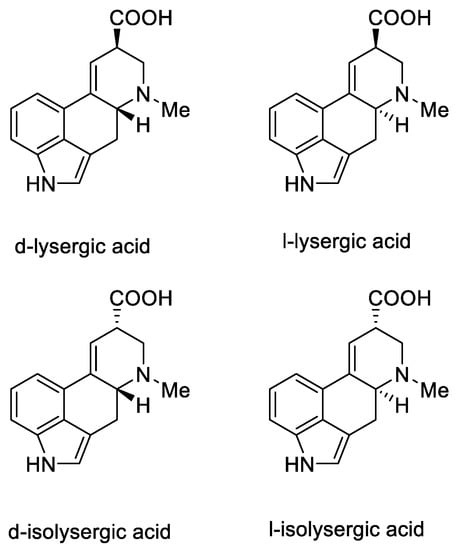
Figure 7. Four stereoisomeric forms of lysergic acid.
Finally, in 1956, at the Lilly Laboratory, the Woodward group was the first to achieve a total synthesis of lysergic acid [30]. In the following years, further structures of ergot alkaloids were identified, and the total syntheses of ergotoxine components were presented.
An explanation of the proposed structures was summarized in the synthetic works of Woodward [30] and Ramage [31]. By examining the pentacyclic styrene derivatives as well as the 2,3-dihydrolysergic acid (+)-butanolamide in the lysergic acid derivatives [32], a conclusion was made about the C-H bonds at C-3 and C-5 being cis and quasi-axial [33].
Surprisingly, not only is the carboxyl/ester group at the C8 position stereoselectively labile, but there is lability at both chiral centers, including at C5, leading to racemization of the system. The balance between structures is easily shifting, e.g., isolysergic acid can be converted to lysergic acid by boiling in methanol or by barium hydroxide in high temperature. Moreover, the same effect can be seen in the natural racemic hydrazine derivatives of lysergic acid [34].
Woodward, who was the first to perform a total synthesis of lysergic acid, proposed a reaction mechanism explaining epimerization at both chiral centers. These postulates were proved spectrally, experimentally, and crystallographically by Ramage two decades later. They proposed that the racemization process was via an achiral tricyclic intermediate (Figure 8), which could be formed by retro Michael fragmentation [35].
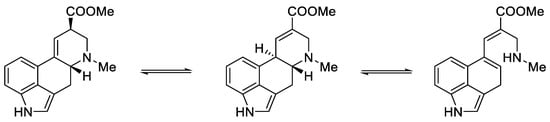
Figure 8. Equilibrium between the forms of lysergic acid, explaining the racemization mechanism.
2. Methods of Lysergic Acid Synthesis
There are many methods of obtaining lysergic acid. They belong to two main types: using biotechnology and synthetic. In total, they allow for the production of 10–15 tons of acid annually [2].
The biotechnological method of obtaining lysergic acid is based on Arthur Stoll’s patent, who in 1918 proposed a method for the efficient separation and purification of ergotamine tartrate from fermented triticale and was put into production by Sandos in 1921. Then, pure lysergic acid can be obtained after amide hydrolysis [36][37].
The second method is proposed by chemists, and concerns the total synthesis of lysergic acid, starting with Kornfeld in 1956 [30]. To this day, 22 methods of synthesis of this compound have been described.
2.1. Woodward’s Strategy
2.1.1. Woodward’s Research Group
The first strategy for obtaining lysergic acid was proposed in 1956 by Woodward’s team [30]. The total synthesis assumed the preparation of the tricyclic ketone 5—1-benzoyl-2,2a,3,4-tetrahydrobenzo[cd]indol-5(1H)-one, then introducing the amine by adding the alkyl fragment at the C4 or C5 position and closing the D ring. The final step involved the isolation of the racemic product—lysergic acid 1 and confirmation of its structure by comparison to the authentic product (Scheme 1).
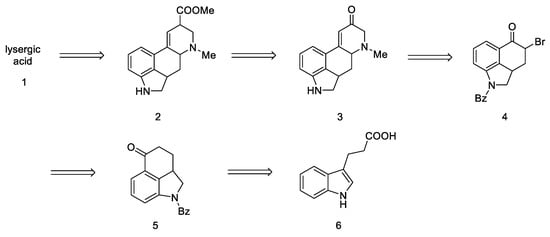
Scheme 1. Retrosynthetic analysis leading to substrate in the Woodward strategy.
Since this is the first work on the preparation of lysergic acid, emphasis has been placed on presenting the line of reasoning described in this article. First, the synthesis of the tricyclic Uhle ketone 8 was presented, and then attempts were made to introduce substituents into it at the C4 and C5 positions. The next section describes attempts to functionalize the ketone by changing it into an unsaturated aldehyde. In the final section, the synthesis leading to the tetracyclic lysergic acid is shown.
Preparation of the starting material. There is a substituted indole system in the lysergic acid skeleton (Figure 9). Indoles are commercially available compounds, and their derivatives can be obtained in the laboratory according to many instructions in the literature.
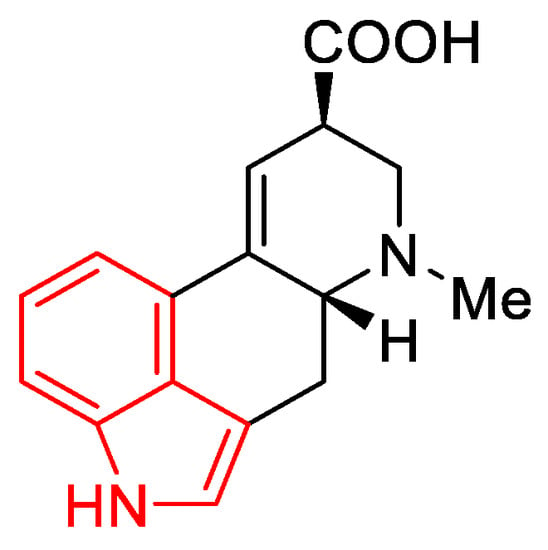
Figure 9. Indole scaffold (red) in the lysergic acid molecule.
The selection of the appropriate indole derivative was not so obvious, however, because the high reactivity of the hetero ring can produce undesirable byproducts based on a more stable structure of naphthalene. The greater stability of naphthalene derivatives is due to the higher resonance energy in relation to the indole [38].
The first step was to functionalize and reduce the starting material to the protected derivative of N-Benzoyl-3-(β-carboxyethyl)-dihydroindole 7 [39]. N-Benzoyl-3-(β-carboxyethyl)-dihydroindole was treated with thionyl chloride to the corresponding acid chloride and then converted into ketone 5 by the Friedel–Crafts reaction. Ketone 5 can be easily hydrolyzed with hydrochloric acid to the free base as well as dehydrogenated over carbon over palladium to the known 3,4-dihydro-benzo[cd]indol-5-one 8, known in the literature as the Uhle’s ketone (Scheme 2) [40].
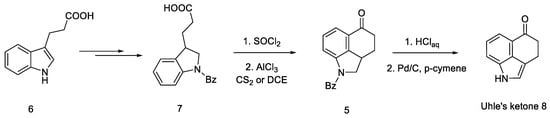
Scheme 2. Obtaining Uhle’s ketone 8.
Very first attempts to obtain lysergic acid. Using the obtained ketone 5, attempts were made to introduce nitrogen into the C4 position (Figure 10).
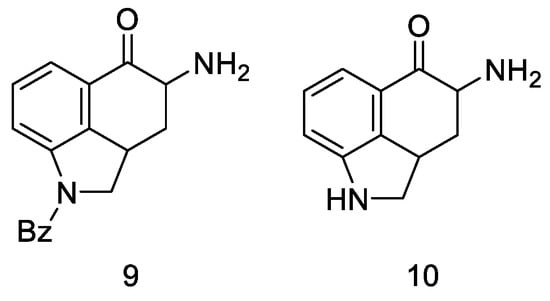
Figure 10. Structural formulas of amine derivatives (9 and 10).
Attempts were made to obtain compounds with free amine or diamine, but all attempts failed at various stages of the synthesis. The reaction of the 4-bromo derivative with methylamine had a complicated course, leading to the naphthalene derivative 14. On the other hand, conversion of ketone 5 to O-toluenesulfonic oxime 15 allowed for obtaining the desired amine 9 (in the form of hydrochloride) by reaction with potassium ethoxide hydrochloride; however, when it was released from the salt, it was very unstable and immediately decomposed, just like diamine. The introduction of the CN and NHCHO functional groups into the C4 position was carried out with low yields; the reaction of oxirane derivatives with amines gave a number of ketal and hydroxyamine derivatives including 12, 13 (Scheme 3).
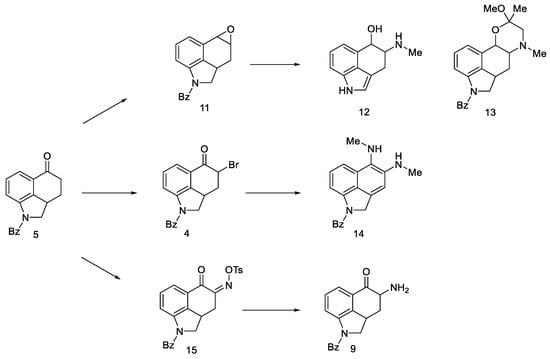
Scheme 3. Derivative products strategies implemented.
Adding a chain at the C5 position was explored as an alternative to introduction of nitrogen at the C4 position. For this purpose, the Reformatsky reaction was carried out with the use of ketone 5. Treatment of bromoacetate in the presence of zinc gave a hydroxyester, which on heating in formic acid gave an unsaturated ester. The ester was converted to the bromoketone 16, which was then reduced to the oxirane 17. After using perbenzoic acid, dioxide 18 was obtained, which, after reacting with the base, gave tetracyclic diol 19 (Scheme 4).
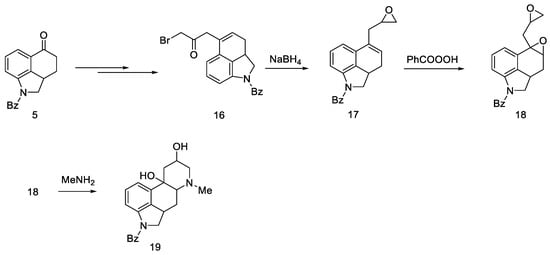
Scheme 4. Diol 19 synthesis.
The obtained product probably could be converted into lysergic acid, but the total yield of nine steps (from ketone 5 to tetracycle 19) was so low that alternative paths were pursued.
A successful synthetic route to lysergic acid. This was achieved by synthesizing the unsaturated aldehyde 22 followed by the preparation of tertacyclic compounds.
This approach was to convert the ketone functional group in 5 into an unsaturated aldehyde 22. To this end, a convergent synthesis leading to its preparation was developed using Darzens reaction [41], and then semicarbazide substitution followed by replacement with a pyruvic acid residue (Scheme 5).
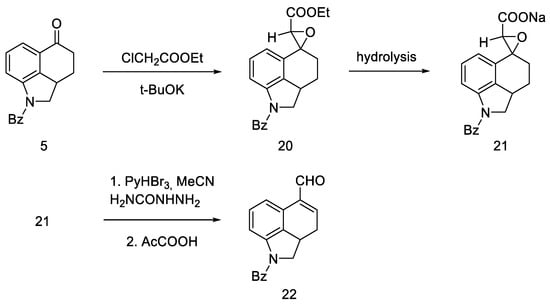
Scheme 5. Aldehyde 22 synthesis.
Woodward proposed three approaches to synthesize an interesting intermediate. One of them turned out to be correct: the aldehyde 22 was converted to oxirane with hydrogen peroxide, then the -CHO group was reduced with NaBH4. Alcohol 23 was reacted with methylaminoacetone–ethylene ketal and oxidized, and the product—ketone 25 (Scheme 6)—was isolated with the desired substituted amino group at C4 position. The disadvantage of this approach is the low yield due to the ketal fragment attachment step.
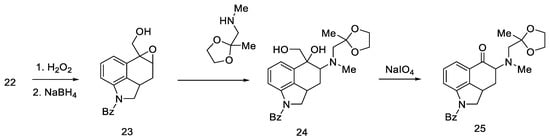
Scheme 6. Ketone 25 synthesis.
However, the reaction of the bromo substitution 4 with methylaminoacetone-ethylene ketal in a nonpolar solvent was tested, and product 25 (Scheme 7) was obtained in two steps from ketone 4 in high yield.
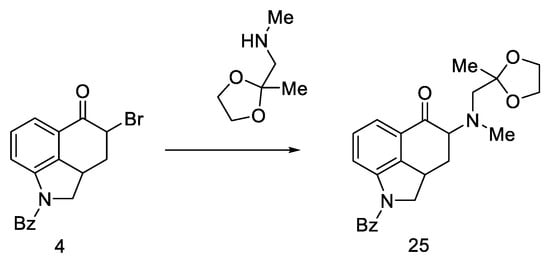
Scheme 7. Reaction of the substitution of bromine 4 with ketal.
The last step involved closing the D-ring, substitution of the -COOH group, and dehydrogenation of the starting dihydroindole structure.
First, protected compound 25 (Scheme 8) was hydrolyzed to a diketone, which was then cyclized. Then, α,β unsaturated carbonyl 30 was reduced with sodium borohydride to allyl alcohol 27.
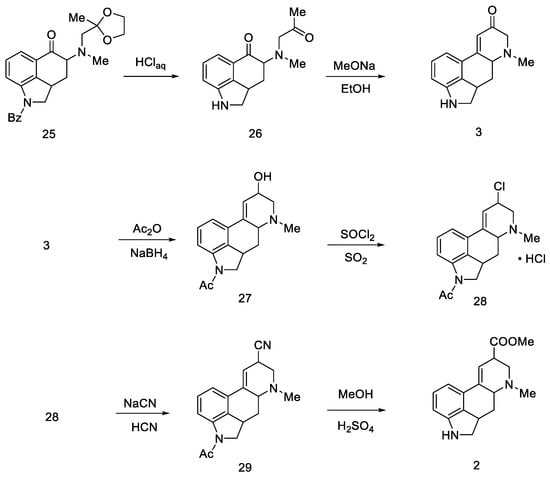
Scheme 8. Woodward pathway to the deprotected key intermediate 2.
Optimization studies were performed according to Price and Krishnamurti, which described the conversion of allyl alcohol to allyl cyanide [42]. It had been found that good results can be achieved by forming chloride 28 followed by nitrile 29 using sodium cyanide in anhydrous liquid hydrogen cyanide. The -COOH group was displaced by methanolysis to an ester, followed by acidic or basic hydrolysis to compound 2.
The final challenge was the oxidation of the B-ring, while maintaining the D-ring double-bond system. For this purpose, catalytic dehydrogenation on the Raney catalyst in the presence of sodium arsenate was used to obtain the target (±)-lysergic acid (Scheme 9) [43].
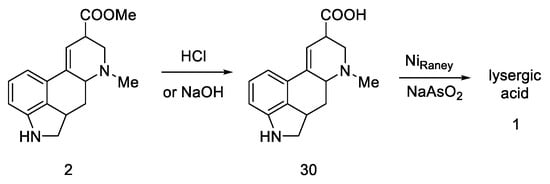
Scheme 9. Woodward pathway to lysergic acid.
Parameters such as melting point, IR and UV spectra, X-ray powder diagrams, pKa, and chromatographic analyses were investigated, confirming the structure of the obtained target product [44][45].
In summary, Woodward’s strategy was based on starting from the indole dihydro derivative, obtaining a tricyclic ketone, introducing nitrogen at the C4 position via ketal, cyclization to the tetracyclic system, hydrolysis of the -CN group, and dehydrogenation of the lysergic acid derivative to the target acid. Final yield based on commercially available substrate was 0.8%.
The next part of this chapter presents syntheses using the key intermediate—hereinafter referred to as the Woodward intermediate (Figure 11), which is the ester 31 (PG—protecting group). It is puzzling that Woodward did not receive the product 31. The name of this intermediate was given by researchers in the following years.
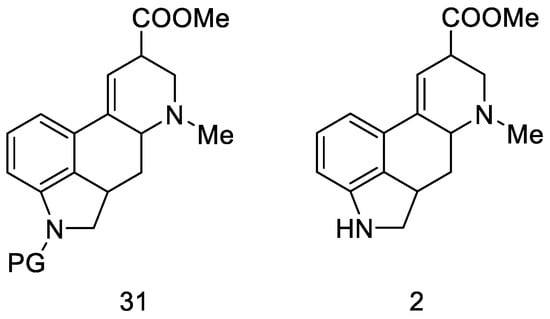
Figure 11. Woodward intermediate 31 and deprotected compound 2.
The synthesis step in Scheme 8 shows the simultaneous methanolysis of the nitrile group and deprotection of the amine, resulting in the product 2 being different from the intermediate 31 by a protecting group. However, the key intermediate can be converted into the product in a manner fully analogous to the procedure outlined by Woodward—lysergic acid 1 can be obtained from it by hydrolysis and dehydrogenation.
2.1.2. Baillarge Group Research
The lysergic acid synthesis performed by M. Julia on the Baillarge team [46] uses the preparation of a tricyclic system obtained as a result of the condensation of 5-bromoisatin 32 and methyl 6-methylnicotinate 33. A key reaction was aryne cyclization. The synthesis was started by obtaining isatin nicotinate 34. Thereafter, reduction of the double bond was performed by treatment with zinc in acetic acid as well as reduction with diborane to obtain a saturated indoline system. The amine was protected by acetylation to give product 35 (Scheme 10).
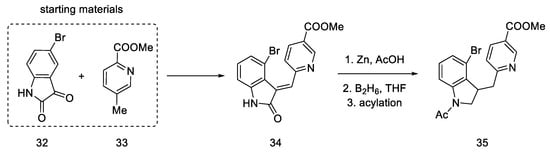
Scheme 10. Beginning of the synthesis proposed by Baillarge team.
In the next step, methyl iodide was added to compound 35, followed by potassium borohydride, whereby the aromatic pyridine ring was reduced to α,β-unsaturated ester 37. It was noted that a mixture of two diastereoisomers differing in physicochemical parameters was obtained. After the use of sodium amide, in ammonia in the reflux, the cyclization reaction was only observed for the (S)-isomer, with a yield of 36%. The product methyl N-acetyl-2,3-dihydrolysergate was isolated, which was in fact an Ac-protected Woodward intermediate (R)-31 (Scheme 11).
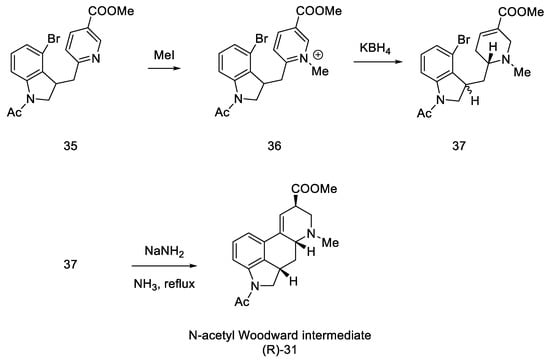
Scheme 11. Synthesis of the protected Woodward’s intermediate.
Using the Woodward and Kornfeld procedure [30][31], this compound can be converted into lysergic acid 1, which formally ends the synthesis. After examining the IR, UV, and mass spectra and preparing a TLC analysis in various systems, to compare the product with the intermediate obtained by Woodward, the identity of the obtained compound was confirmed.
2.1.3. Ramage’s Research Group
R. Ramage, W. Armstrong, and S. Coulton, in 1981, used and improved Woodward’s strategy [31]. Optimization of the synthesis of tricyclic aldehyde 22, which was then a substrate, in order to obtain Woodward’s intermediate 31, was performed. A procedure was proposed, which consisted of passing through the Wittig reaction to the unsaturated diester 38 and then introducing the amino group using the Curtius degradation procedure and cyclization of the D ring.
In the work of the Ramage group, a lot of emphasis was placed on structures and intermediates through the analysis of NMR spectra, which confirmed the reaction mechanism proposed in the first synthesis. In addition to NMR studies, HPLC chromatogram was also investigated as were the crystallographic analyses and UV/IR spectra of the products. Thanks to these studies, it was found that lysergic acid formed as two epimers, and their ratio was determined.
Aldehyde 22 (having a well-described, two-step synthetic pathway via semicarbazide in the experimental part) [31], was converted with a Wittig reaction through resonance-stabilized ylide into an unsaturated diester 38. The reaction is stereospecific (configuration E), because the Wittig reaction with resonance stabilized ylides leads to systems having a carbonyl trans function to a larger group at the beta position [47]. Then, chemoselective hydrolysis of the ester bond of the -COOtBu group was carried out, and the Curtius degradation procedure was optimized (the total efficiency of this procedure was 80% [48]); at the beginning, tetramethylguanidine azide and mixed diphenylphosphinic anhydride were added to a diester 38, and thermal rearrangement took place. Azide to the isocyanate 39 was performed, followed by hydration and decarboxylation to the amine. To avoid side reactions, it was decided to control the hydration using tosylic acid monohydrate. Thus, the amino group was introduced into compound 40 as a protected tosyl salt (Scheme 12).
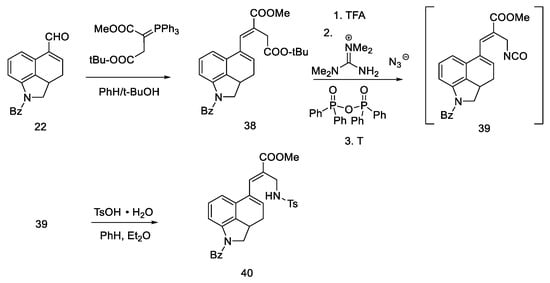
Scheme 12. First part of the Ramage synthesis.
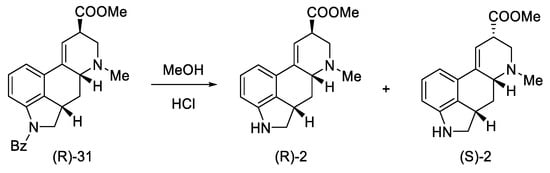
Secondary amines have been shown to cyclize much more easily than primary amines [49]. The Woodward reaction mechanism model also assumes that the amine is an intermediate, spontaneously cyclizing to a 2,3-dihydrolysergic system [32][50]. Therefore, the Eschweiler–Clarke reaction [51][52] was performed with HCOOH/HCHO, yielding three tetracyclic products ((R)-31, (S)-31, 41) in a 9:3:2 ratio, from which two Woodward intermediate epimers (R/S)-31 were separated. The use of boiling methanol allowed for the epimerization of the isolysergic acid related compound (S)-31 to the lysergic derivative (R)-31 (Scheme 13).
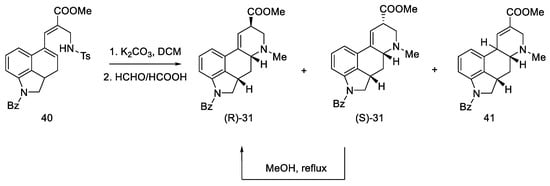
Scheme 13. Second part of the Ramage synthesis.
Pure compound (R)-31 was treated with HCl in MeOH to yield debenzylated epimeric lysergic acid derivatives (R)-2 and (S)-2 in a 5:2 ratio (Scheme 14). These products were identical to the material used in the first Woodward lysergic acid synthesis [30]. As a result of further work of the team, derivatives were also obtained, which were secondary and not tertiary amines in the D ring, enabling their further functionalization towards interesting derivatives.
Scheme 14. Obtained epimers of dihydrolysergic acid derivatives 2.
Treating the Curtius procedure as a single one-pot reaction, the transition from ketone to Woodward intermediate consists of four reactions [53].
2.1.4. Rebek’s Research Group
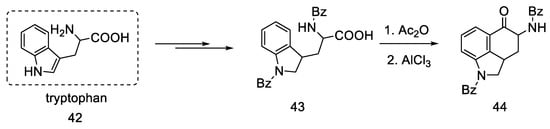
Rebek’s team proposed a different, simple starting material for the preparation of lysergic acid, which is the natural amino acid tryptophan 42 [54][55]. The first part involved obtaining the tricyclic ketone, analogous to the Woodward path [30]. Amino acid derivative 42 was converted to ketone 44 by adding acetic anhydride and aluminum chloride (Scheme 15).
Scheme 15. Preparation of the Rebek substrate 44.
The obtained ketone is functionalized by using the starting amino acid—it has an amino fragment in the C4 position.
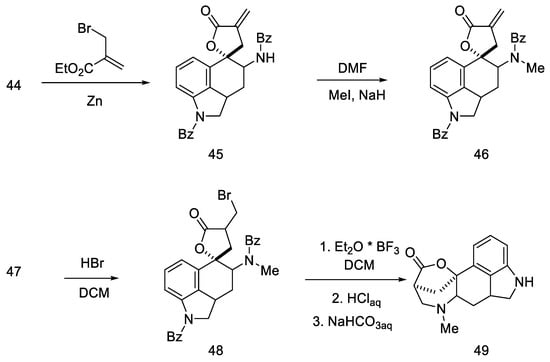
The next steps involved closing the D ring. Initially, ketone 44 was converted into spiro-lactone 45, giving methylated compound 46 in the presence of sodium hydride and methyl iodide, which was hydrobrominated and then closed in the three-stage Henessian sequence [56] into the pentacyclic structure of the lactone 49 (Scheme 16).
Scheme 16. The first part of Rebek synthesis.
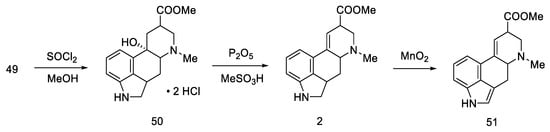
The last part was the formation of an ester group and dehydration to the lysergic acid structure. Lactone 49 was opened with thionyl chloride to dihydrochloride ester 50, which was dehydrated to olefin 2. Oxidation under mild conditions made it possible to obtain a mixture of deprotected isolysergic and lysergic acid esters 51 (Scheme 17), the physicochemical parameters of which were consistent with the data in the literature [57].
Scheme 17. The second part of Rebek synthesis.
2.1.5. Kiguchi’s Research Group
The Kiguchi group proposed, in 1982, the synthesis of the Woodward intermediate using the photochemical cyclization reaction of the D ring [58].
This work is an interesting attempt to obtain an optically pure intermediate. The original Woodward article [30] did not focus on obtaining pure chiral products. Instead, the lysergic acid that formed was a racemic mixture. The chirality of the intermediates was not tested either—as in Rebek’s work [54], a mixture of four final ester stereoisomers was obtained. Therefore, this procedure is linked to the Ramage article [31].
The synthesis started with a more complicated structure than proposed by Woodward or Rebek—with the tricyclic ketone (benzo(f)quinoline derivative) described in the literature 52 [59][60]. Known ketone 52 was treated with methylamine to give imine 53 as an intermediate, which was easily converted to enamide 54 after acylation with 3-furoyl chloride in the presence of triethylamine. Reductive photocyclization was performed, leading to lactam 55 (Scheme 18).
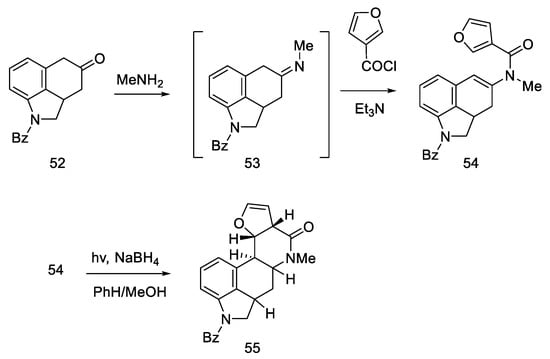
Scheme 18. The first part of Kiguchi synthesis.
Reduction of lactam 55 with LiAlH4 followed by reprotection of the amine gave a mixture of two diastereoisomers 56 (Scheme 19). The decarbonylated amine (R)-56, having the stereochemistry corresponding to the d-(+)-configuration of lysergic acid, was isolated by crystallization.
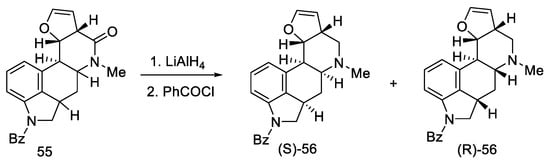
Scheme 19. The second part of Kiguchi synthesis.
Oxidation of amine (R)-56 gave glycol 57, which is a diastereomeric mixture of two cis-glycols. Without further purification, oxidation of the glycol with sodium periodate gave a mixture of epimeric hydroxyaldehydes 58 (Scheme 20) [58].
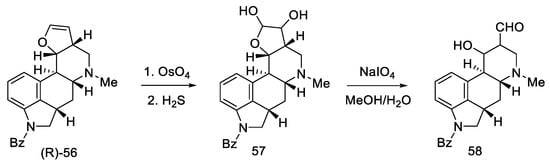
Scheme 20. The third part of Kiguchi synthesis.
After oxidation of hydroxyaldehydes to hydroxyesters, it was investigated that the products contain two epimers, which are precursors of isolysergic (S)-59 and lysergic (R)-59 acids. Dehydration of both hydroxyesters gave one product—unsaturated ester (R)-31, which is an optically pure Woodward intermediate (Scheme 21). This selectivity is due to the higher thermodynamic stability of β-equatorial esters relative to the α-axial esters [32][50]. Spectral examination confirmed the identity of the structure to the compound obtained by Ramage [31] and Woodward [30].
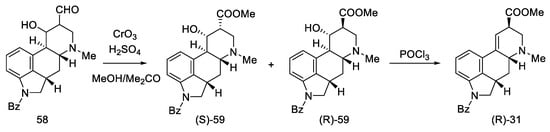
Scheme 21. The last part of Kiguchi synthesis.
Continuing the work of the team, during the first synthesis of (±)-isofumigaclavine B in 1985 [61], the synthesis of lysergic acid was almost completed. Removal of Bz protecting group and oxidation of mixed epimers of ester (31) produced lysergic acid ester 51 (Scheme 22).
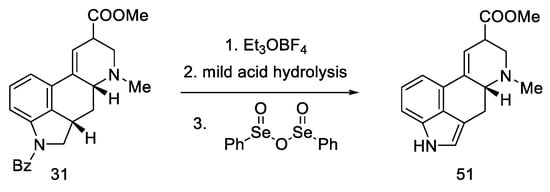
Scheme 22. Additional Kiguchi research.
The use of the photochemical cyclization reaction is an interesting alternative to the previous proposals for the closing of the D-ring. It is a completely fresh approach, using a different branch of chemical synthesis. This work was an important step towards obtaining pure chiral d-lysergic acid.
According to the latest reports, structure 51 can also be obtained in three–five steps from a nicotinic acid ester 60 (Scheme 23) [62].
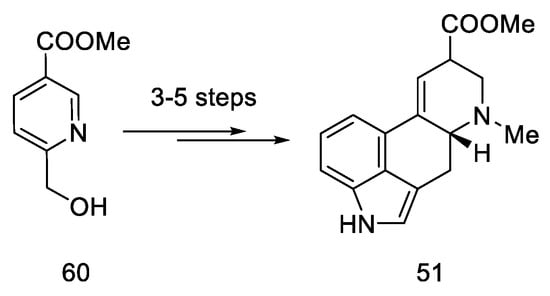
Scheme 23. A new method for the synthesis of compound 51.
2.2. Hendrickson’s Strategy
2.2.1. Direct Hendrickson’s Research
Hendrickson’s group approached the topic of lysergic acid synthesis from a new angle. Using readily available components such as an indole halide and a pyridine derivative, they obtained the lysergic acid structure using a coupling reaction followed by cyclization of the aldehyde to give the D ring—the key Hendrickson intermediate [63]. The desired product was then obtained by reduction with a classical borohydride. The main advantage of this approach is the convergence and simplicity of the method, as well as the total efficiency. The product was obtained in eight steps, and the yield was 10.6%.
The assumptions of the strategy proposed by Hendrickson are: synthesis of lysergic acid through the shortest possible path, without protection of the indole fragment, and introduction of the D ring through Suzuki coupling.
The synthesis was started with the preparation of the halogen derivative of indole 61, which was converted into compound 62. Starting from the dicarboxylic derivative of pyridine 63, the synthesis of its N-oxide was performed, and then, with thionyl chloride, it was converted into a chlorine derivative of 64 (interestingly, only under these conditions the meta derivative is obtained, instead of the classic ortho/para substitution). The aromatic substrates, thus prepared, were easily coupled via a palladium-catalyzed Suzuki reaction to the tricyclic derivative 65 (Scheme 24). An analogous coupling was simultaneously described by Doll [64].
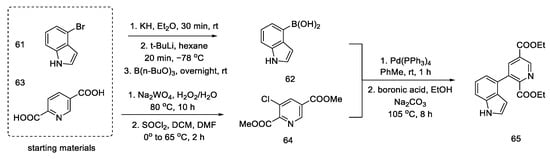
Scheme 24. Coupling reaction leading to a tricyclic system 65.
Closing the tetracyclic system posed some difficulties—diester 65 did not give the expected cyclization product; therefore, the ester was reduced to alcohol and then gently oxidized to active aldehyde 66, which, under basic conditions, resulted in a tertiary cyclic alcohol 67, known as Hendrickson’s intermediate (Scheme 25).
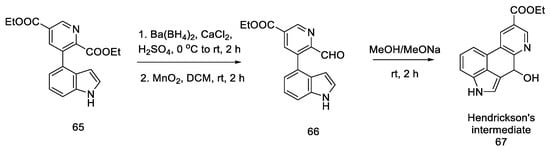
Scheme 25. Obtaining Hendickson’s intermediate.
Reduction of the alcohol 67, N-methylation of the pyridine derivative and reduction of the aromatic system were performed to obtain a mixture of 69: lysergic acid ester and isolysergic acid ester in a ratio of 6:1 (Scheme 26). The last three steps were performed without isolation to avoid losses due to product instability. Using the procedure from the literature on the isomerization of the iso-lysergic acid form, it was converted to the desired acid under basic conditions. Its physicochemical properties such as melting point were checked to confirm its structure.
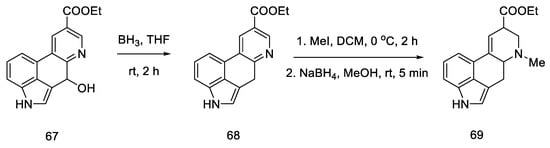
Scheme 26. Synthesis of lysergic acid ester proposed by Hendrickson.
However, this pathway of obtaining lysergic acid was proven to be not feasible, as described later in this chapter.
2.2.2. Problems Noticed by Bekkam
In 2011, Bekkam, Mo, and Nichols [65] repeated the synthesis of lysergic acid using the conditions proposed by Hendrickson [63], using methyl esters 70. Surprisingly, it turned out that the cyclization step to compound 67, as shown in Scheme 25, did not occur.
The reaction was attempted multiple times; neither the change of researcher nor the multiple repetitions of the reactions with different amounts of base, different time, and different temperatures resulted in the desired effect. The obtained compound 71 was the product of the cyclization and aromatization of the system (Scheme 27).
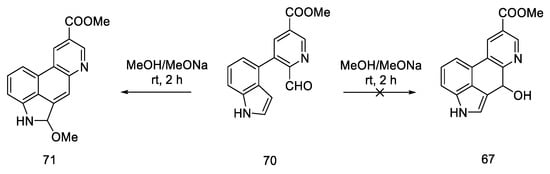
Scheme 27. Structural formula of the product obtained in the literature procedure repeated by Bekkam.
The structure was unambiguously checked and determined by melting point and the analysis of one- and two-dimensional NMR spectra (1H, 13C, COSY, HMBC, TCOSY, HMQC).
The reason for obtaining the other main product is the resonance stabilization of the indoquinoline system, as well as the fact that the dihedral angle between the carbonyl carbon and the C3 carbon is 160°, which allows for easy elimination under basic conditions according to the E2 mechanism.
Researchers have not been able to contact members of Hendrickson’s group, but it must be assumed that this particular path is not feasible.
However, before these problems were noticed by Bekkam, Hendrickson’s strategy was used to formally synthesize lysergic acid by Yigang Zhao [66] and Beaundry [67].
2.2.3. Yigang Zhao Work
Yigang Zhao’s doctoral dissertation from the Snieckus group presents an alternative method of obtaining the key Hendrickson intermediate [66][68]. Selective amidation, DoM (Directed Orto Metalation-Suzuki cross coupling) cross coupling, and the chemoselective reduction of an amide to an aldehyde using Schwartz’s reagent were used for this purpose. This simplifies Hendrickson’s strategy by eliminating one step. Then, according to the reactions in the literature, the compound can be deprotected and cyclized [63].
The first step was selective amidation and transesterification to 73. The substrate prepared in this way can be coupled using the conditions optimized by the researchers for a tricyclic system 74 (Scheme 28). The reduction step with the use of an organozirconium reagent took place after protection of the indole N-H; therefore, Boc-protection was proposed.
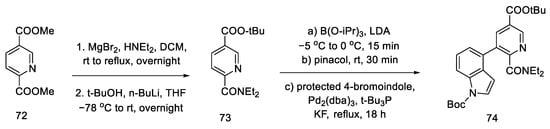
Scheme 28. Coupling reaction leading to a protected tricyclic system 74.
Carrying out the reduction with the Schwartz reagent allows for obtaining the aldehyde in an efficient manner, even in the presence of a more reducible ester. It was postulated (dotted arrow) that in this way, after carrying out two steps, it could lead to the Hendrickson’s intermediate 67 (Scheme 29).
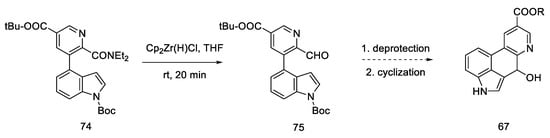
Scheme 29. Synthetic path leading to the Henrickson’s intermediate, as postulated by Yigang.
This modification makes it possible to obtain the intermediate in six, instead of seven steps, but it involves the use of organometallic reagents such as BuLi and Cp2Zr(H)Cl.
2.2.4. Beaundry’s Research
During their research, Beaundry’s group proposed the formal synthesis of lysergic acid via the Hendrickson intermediate as an example of using this method for the preparation of substituted indoles [67]. The synthesis relies on the coupling of a terminal alkyne alcohol and a halogen derivative of the pyridine ester 76 and the substitution of an amine with a cyclic substituent, followed by the closure of the indole system, leading to the known precursor 65, from which the Hendrickson’s intermediate can be obtained in two steps.
Chloropyridine 76 was prepared according to the procedure in the literature [63]. The Sonogashira coupling of the alkyne and chloropyridine was performed using microwave heating. Compound 77 was then activated with triflate anhydride and converted to a secondary amine 78 (Scheme 30).
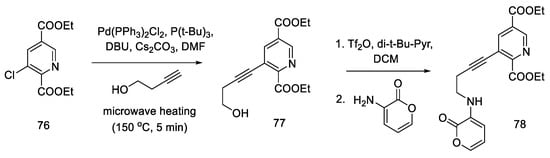
Scheme 30. Beginning of the synthesis proposed by Beaundry.
The research of the Beaundry group focused on the preparation of the bicyclic system of substituted indole, and, as a result, the methods of oxidative cyclization were optimized: in the first step, butyl nitrile was added under basic conditions and strongly heated with microwave, and then HO-TEMPO was added in the presence of copper (I) salt and controlled amounts of oxygen to give the indolyl-pyridine 65 in good yield.
Hendrickson’s intermediate 67 can be obtained in two steps, by the reduction and cyclization described above (Scheme 31) [63].
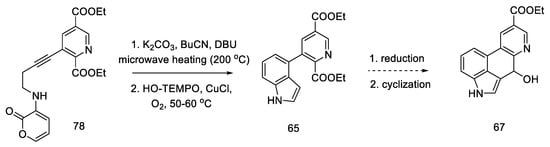
Scheme 31. Ending of the synthesis proposed by Beaundry.
2.3. Szantay’s Strategy
2.3.1. Direct Szantay’s Research
Szantay proposed the synthesis of lysergic acid using an aldol condensation reaction to close the D ring. The first presentation of the synthesis of (+)-lysergic acid is also a great achievement, thanks to the use of the obtained tetracyclic system, which was separated by crystallization with a chiral auxiliary. In doing so, he obtained a key intermediate (R)-82 (Szantay’s intermediate). The next four steps are required to obtain lysergic acid. The article also describes, in addition to the lysergic acid total synthesis, the synthesis of an important derivative of ergocriptinine, which is a peptide derivative of lysergic acid on a large scale [69].
The synthesis involved going through the following steps: obtaining a tricyclic ketone, closing the fourth ring using the aldol reaction, enantioselective separation of the obtained product, and substituting the carbonyl group with a carboxylic acid to obtain (+)-lysergic acid.
The synthesis was started with the preparation of the bromo derivative of Uhle’s ketone 79. The original procedure for the preparation of Uhle’s ketone assumed starting from the 3-indolpropionic acid 6 by the Goto method [70]; however, only the preparation of the bromo derivative made it possible to carry out the amination reaction. The authors proposed two methods of obtaining compound 80 from the bromo derivative 79 (Scheme 32 shows a two-step synthesis with a yield of 35%, but an alternative is a four-step process with a total yield that is higher by 4%). Next, the NH group of the indole was deprotected.
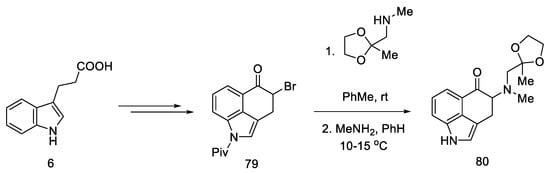
Scheme 32. Beginning of the synthesis proposed by Szantay.
The next step involved closing the D ring. For this purpose, intramolecular aldol condensation was employed, using 1,5-dicarbonyl moiety 81, formed after the deprotection of ketone 80. However, selecting the optimal conditions was a difficult task. Bases ranging from tert-butoxide to bases such as LiHMDS were tested with no effect. A satisfactory effect was achieved using the Eschenmoser conditions—LiBr and triethylamine [71].
The authors suggest that the tetracyclic product 82 is formed by lithium bromide activation of the carbonyl groups in the presence of an amine, due to the higher affinity of lithium for oxygen than nitrogen. The presence of the amine is essential—it is used to abstract a proton from the alpha position next to the carbonyl group. These initial synthesis steps make use of the synthesis intermediates already proposed by Woodward [30].
The best effect was achieved by performing the deprotection and cyclization reaction without isolating compound 81 and isolating compound 82 by crystallization to give a total 60% yield of these steps (Scheme 33).
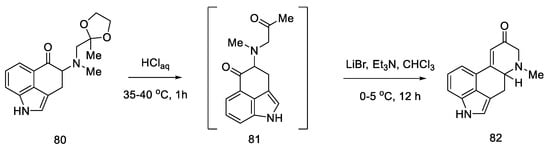
Scheme 33. Preparation of the carbonyl α,β-unsaturated system 82.
Enantioselective separation was performed by crystallization with chiral dibenzoyltartaric acid, and pure compound (R)-82 was obtained, contributing to the first asymmetric synthesis of (+)-lysergic acid. The physicochemical properties of the obtained compound were compared with the degradation product of natural (+)-lysergic acid. The optically pure isomer (R)-82—Szantay’s intermediate—was reacted with isonitrile to produce a tosylated formamide derivative 83 (Scheme 34).
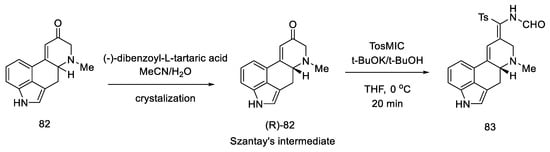
Scheme 34. Obtaining Szantay’s intermediate.
Basic hydrolysis gave the R/S nitrile mixture (84), which was converted to the R/S methyl ester mixture 51 by Pinner reaction. Basic ester hydrolysis gave pure (+)-lysergic acid by epimerization (Scheme 35) [69].
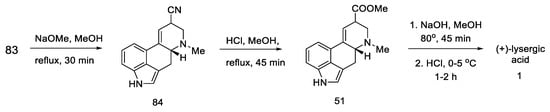
Scheme 35. Synthesis of lysergic acid proposed by Szantay.
2.3.2. Continuation of Szantay’s Research
A year later, in 2005, Helvetica Chimica Acta published a review presenting the entire range of works by Szantay’s group [72]. Unfortunately, the only successful attempt to obtain lysergic acid was the synthesis presented in Section 2.3.1, although the work resulted in a number of interesting four- and five-cyclic compounds based on the ergoline structure, obtained by Stobbe (Figure 12).
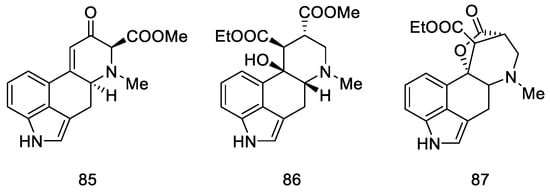
Figure 12. Structural formulas of compounds obtained in further works of Szantay’s research group.
2.3.3. Garner’s Research
In 2021, Garner and Rathnayake published a study that used Szantay’s intermediate to obtain lysergic acid [73]. This is a novel approach that involves carrying out an intramolecular cycloaddition reaction of an azomethine ylide, generated in situ, followed by ring expansion to Szantay’s ketone. During these reactions, C and D’ rings (where D’ is a five-membered precursor of the D ring in the ergoline structure) are obtained, and then they are transformed into CD rings. The disadvantage of this method is a very large number of stages—Szantay’s intermediate (R)-82 is obtained in 16 stages, with a total yield of 1.0% (average—75% per step).
The synthesis was started with 4-bromoindole 61, which was converted into the bromoindole 88 derivative, according to a procedure in the literature [74]. Then, using a Sonogashira coupling and removal of the TMS group, terminal acetylene 89 was obtained, which was substituted with the sulfonyl group. The obtained compound 90 was deprotected to the form of an alcohol and oxidized to aldehyde 91, which was a substrate for the intramolecular cycloaddition reaction (Scheme 36).
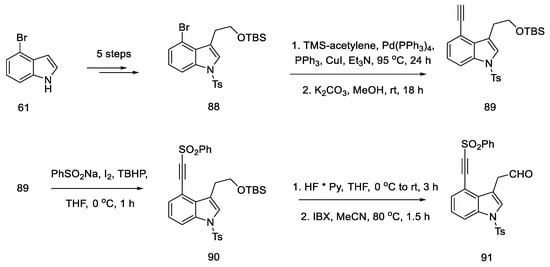
Scheme 36. Beginning of the synthesis proposed by Garner.
The second substrate was compound 93, a chiral glycylsultam, obtained in a five-step synthesis from (2S)-bornane-2,10-sultam 92, a camphor derivative. The steps of substituting the acid halide with an organolithium agent were performed, followed by the introduction of the -NH2 group by the azide. The intramolecular cycloaddition of an azomethine ylide is an asymmetric cycloaddition reaction of three elements: aldehyde, dipolarophilic factor (in this case in one substrate 91), and glycylsultane. As a result of the reaction—as the authors suggest—through the intermediate 94, a system of four ABCD rings is formed (Scheme 37) in an enantioselective reaction.
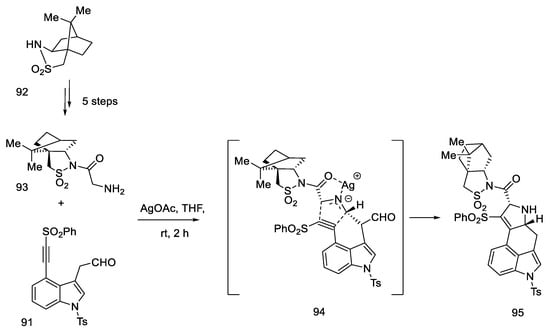
Scheme 37. Preparation of a tetracyclic derivative 95 with a camphor substituent.
In the following steps, a fragment of the camphor derivative was removed, and the D ring substituent was reduced, yielding alcohol 97. Then, using the five- to six-membered ring expansion reaction proposed by Cossy and Charette [75][76], the correct system was obtained 98.
The oxidation and deprotection of compound 99 allowed for obtaining Szantay’s intermediate (R)-82, from which the proper, optically pure (+)-lysergic acid can be obtained in four steps (Scheme 38).
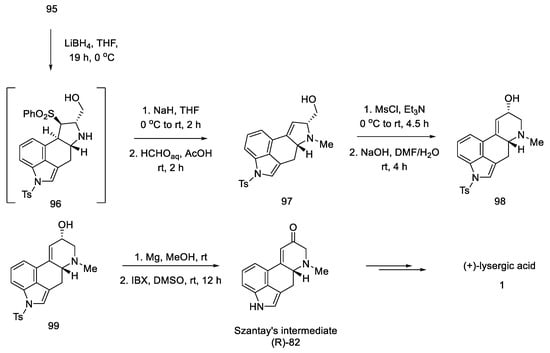
Scheme 38. Synthesis of lysergic acid proposed by Garner.
2.4. Closing the C/D-Ring Using the Heck Method
The Mizoroki–Heck reaction, including the intramolecular version, is the reaction of the selective coupling of an aryl or vinyl halide and an alkene in the presence of a palladium catalyst under basic conditions [77]. It is often used to enclose the six-membered C-ring in the ergoline structure. A lot of synthetic work was based on the scheme: preparation of halogen-indole or tryptophan derivatives, then coupling of the D-ring, manipulation of substituents to create favorable conditions for the intramolecular Heck reaction, and conversion of the functional groups to (+)-lysergic acid.
2.4.1. Ortar and Kurihara Groups
Strategy of closing D-ring using the Heck method. In 1988, the Ortar group was the first to propose the preparation of lysergic acid through the palladium-catalyzed Heck coupling [78]. The starting material in the synthesis was Uhle’s ketone derivative 5 [30].
The tricyclic olefin 100 was obtained, as a substrate for the coupling reaction, after the use of a pyridine derivative and triflic anhydride. The second substrate was the unsaturated amino ester 102, which was obtained by carbanion generation from amino ester 101, the addition of formaldehyde, the introduction of mesyl chloride, and elimination promoted by strong base DBU.
Heck coupling resulted in compound 103, which readily cyclizes in the presence of hydrochloric acid (Scheme 39).
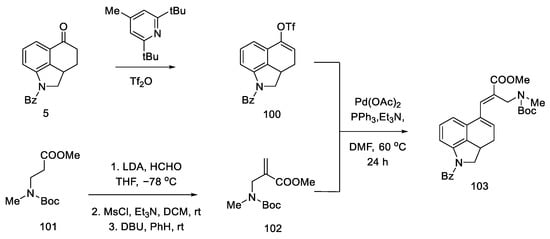
Scheme 39. Preparation of a tricyclic system by Heck coupling as the source of the D-ring.
A mixture of the two epimers 31 was obtained with a yield of 60%, in a ratio of 1.7:1. Despite being consistent with the spectral data reported by Ninomiya and Rebek, refluxing the ester mixture 31 in methanol did not result in a 3:1 ratio, as reported in the literature [53][57]. Nevertheless, using the obtained mixture of esters, the target product can be prepared, which ends the formal synthesis of lysergic acid (Scheme 40) [30][31].
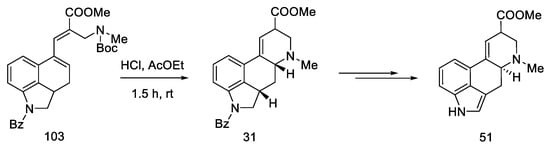
Scheme 40. Synthesis of lysergic acid ester 51 proposed by Ortar.
Kurihara group research. Two years earlier, the Kurihara group proposed a method of obtaining lysergic acid using the same substrates as Ortar, reaching the same analogous intermediate, which can be cyclized; however, in this case, a ring-closing reaction was performed without using the Heck coupling method [79][80]. The same intermediate that was already described by Woodward is obtained [30].
The substrate for the proposed synthesis was the Uhle ketone derivative 5 [30]. Converting the carbonyl functional group first to nitrile and then to aldehyde via DIBAL-H reduction gave compound 22. Then, using a strong organolithium base, LDA, introduction was performed into the tricyclic structure of the amine-ester moiety.
In the next stage, the alcohol was changed to the -OMs group, obtaining the mesylate 104 (Scheme 41).
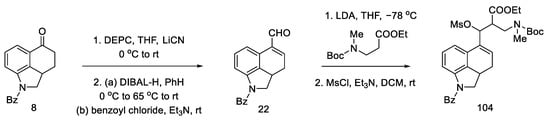
Scheme 41. Preparation of a tricyclic system by Kurihara, analogous to that of Ortar.
Conversion into Woodward’s key intermediate ethyl ester could be carried out in two ways: either 104 was deprotected first and then cyclized, or it was first treated with DBU, and, in the next step, the protecting group was removed. The reaction runs through intermediate products, which was used by the Ortar group [78]. Three products were obtained: a mixture of (R) and (S) of the expected N-benzoy1-2,3-dihydrolysergate and also the double-bond isomerization product. Recrystallization of the mixture in ethyl acetate allowed for the isolation of the pure product. The last step was hydrolysis and re-esterification, leading to the known compound (R)-31, from which, according to the literature [61], lysergic acid can be obtained (Scheme 42).
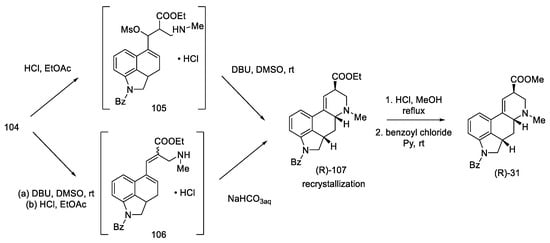
Scheme 42. Preparation of a lysergic acid protected ester (R)-31 by the Kurihara strategy.
2.4.2. Fukuyama’s Research
Strategy I. In 2009, the Fukuyama group proposed the synthesis of (+)-lysergic acid [79], which so far had only been possible through the Szantay approach [72]. Lysergic acid was prepared by closing the C-ring via an intramolecular Heck reaction using the tricyclic compound 117. In turn, this compound could be obtained from the bromomagnesium indole derivative and from the disubstituted tetrahydropyridine. Chiral tetrahydropyridine, the structure of which corresponds to the D-ring, was obtained from L-pyroglutamic acid by a dissrotational electrocyclic reaction [81].
The synthesis began with the acylation of lactam 108 and the reduction and dehydration of the hemiaminal to enamide 109. Using catalysis and two-phase conditions, alkene was converted to dibromocyclopropane 110, which rearranged at high temperature into carbocation 111 and then into the substituted dihydropyridine 112 (Scheme 43).
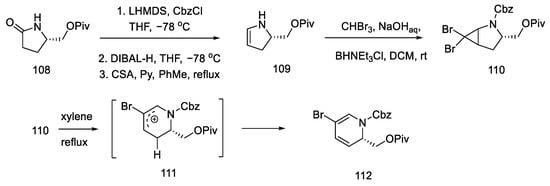
Scheme 43. Synthesis of dihydropyridine 112.
As a result of the next seven stages of the synthesis, a tetrahydropyridine acid chloride 115 was prepared, which could be subsequently coupled to a tricyclic system using an organometallic compound. The resulting compound 112 was reduced and then carbonylated under catalytic conditions. Using a Ritter’s method [82], the Cbz protecting group was replaced with a -Ns group (Scheme 44). Then, it was possible to apply the Jones oxidation with chromium oxide, and the compound obtained in this way was transformed into acid chloride 115 by the Ghosez procedure [83]. Interestingly, this is the first example of the synthesis of optically pure dihydropyridine from L-pyroglutamic acid. As a result, a chiral center is introduced into the compound, which makes it possible to obtain pure (+)-lysergic acid.
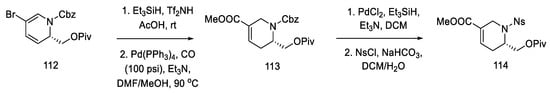
Scheme 44. Protection of the tetrahydropyridine with a nosyl group.
The coupling of the D-ring derived from piperidine 115 and the 4-bromoindole derivative was performed in moderate yield. The nitrogen in the indole was protected with Alloc, whereby the carbonyl group was activated and reduced to a diastereomeric mixture of alcohol 117 (Scheme 45).
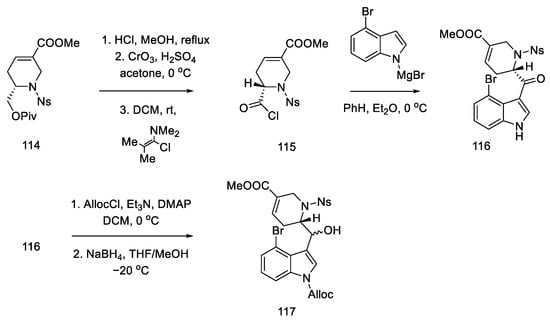
Scheme 45. Synthesis of tricyclic alcohol 117.
Subsequent reactions involved the deprotection and methylation of tetrahydropyridine nitrogen to give alcohol 118. Next, indole nitrogen was deprotected, and the alcohol was reduced with triethylsilane under acidic conditions. The indole nitrogen was -Boc protected, and the double bond was isomerized with 2,6-tert-butylphenol and lithium tetramethylpiperide to give a mixture of diastereomeric esters 120 (Scheme 46).
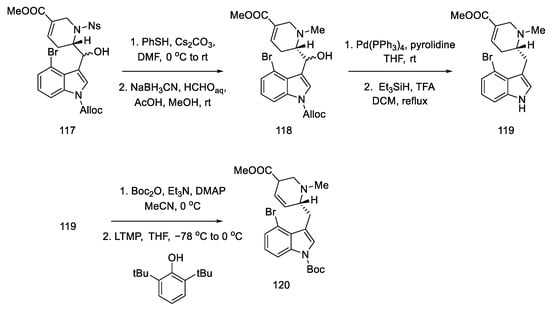
Scheme 46. Preparation of ester 120.
These isomers were separated and subjected to an intramolecular Heck reaction closing the C-ring of the ergoline structure to obtain the protected ester. The desired trans isomer was obtained in excess (3:1). After deprotection of the indole nitrogen with trifluoroacetic acid, ester 121 was obtained, which can be converted to lysergic acid (Scheme 47) [84].
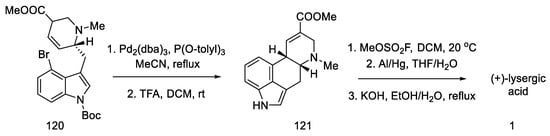
Scheme 47. Preparation of lysergic acid in Fukuyama strategy I.
Despite the undoubted advantage of the approach proposed by Fukuyama, which allows for obtaining pure (+)-lysergic acid, there are many disadvantages associated with it. First of all, the number of stages, since the procedure involves over 30 steps from the starting lactam to the final product. Additionally, the use of a large number of protecting groups should be mentioned as a consequence of the use of numerous organometallic compounds, such as DIBAL-H, LTMP, or Grignard compounds, and other requirements related to such active reagents, for example temperature control. Realizing the disadvantages of the proposed approach, Fukuyama introduced numerous alterations in subsequent publications.
Strategy II. The Fukuyama group continued to work on new methods of obtaining lysergic acid. The aim was to simplify and shorten the synthesis and increase the overall yield. A synthesis based on a one-pot double-cyclization reaction, simultaneously closing the B and C rings, thanks to an intramolecular amination reaction to the aromatic system (to the indole AB system) and the palladium-catalyzed Heck reaction (ring C), have been proposed [85]. In order to obtain the substrate for the double-cyclization reaction, the synthesis of compound 131 was carried out by the coupling method of dibromo-iodobenzene and nitrated olefin 127. The synthesis uses 2-(but-3-en-1-yl)-propane-1,3-diole 122 as a starting material.
The synthesis of the optically active nitroolefin 127 was started with lipase-mediated acetylation and conversion of the second -OH group to the amide under Mitsunobu conditions, achieving an optically pure product (95% ee) and preventing racemization. The cyclic acetate 124 was then obtained after dehydration of the hemiaminal (Scheme 48).
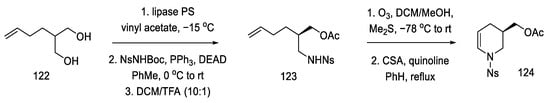
Scheme 48. Synthesis of cyclic acetate-enamine 124.
Acetate 124 was converted to the corresponding p-nitrobenzoate enamide 125, which was recrystallized from methanol. Enamide 125 was then treated with NIS in methanol, followed by dehydrogenation with DBU to give methoxy-olefin 126. Stereoselective attachment of the side chain was performed by treatment with allyltrimethylsilane (Scheme 49).
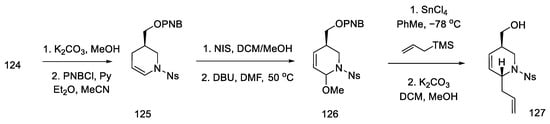
Scheme 49. Introducing an allylic system into molecule 126.
After modifying the protecting groups, nitroolefin 127 was obtained in two steps by reacting the formed aldehyde with nitromethane (Scheme 50).
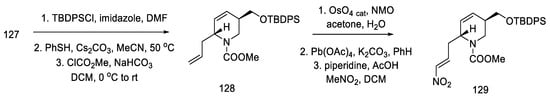
Scheme 50. Preparation of nitroolefin 129.
By using nitroolefin 129, the key cyclization reactions leading to the tetracyclic ergoline structure could be performed. Synthesis was continued with the formation of an organolithium compound from dibromoiodobenzene and coupling it with a nitroolefin to form the bicyclic compound 131. Then, using the Heck coupling and intramolecular amine cyclization, two new rings—B and C—were formed, yielding structure 132 (Scheme 51).
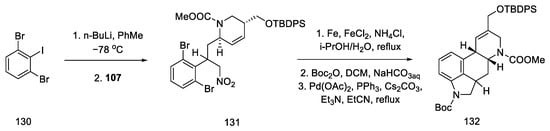
Scheme 51. Forming the B and C rings in Fukuyama strategy II.
The next steps were the manipulation of the protecting groups to give the diester 134 and migration of the double bond from the C8–C9 position to the C9–C10 position using DBU (Scheme 52) [86].
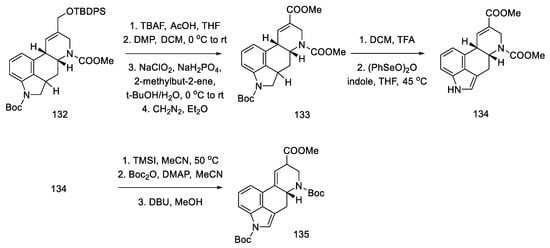
Scheme 52. Preparation of a protected lysergic acid ester 135 by Fukuyama strategy II.
Finally, gradual deprotection of both Boc groups and the reductive methylation of dehydropiperidine yielded lysergic acid methyl ester 51 as a mixture of diastereoisomers, with spectroscopic data that were identical to those described in the literature [69]. This compound can be converted into (+)-lysergic acid, according to the procedure proposed by Szantay (Scheme 53) [69].
Scheme 53. Deprotection and methylation of lysergic acid ester 51.
In conclusion, an asymmetric synthesis of the (+)-lysergic acid ester was achieved using the Heck coupling reaction to close the C-ring of the ergoline structure. The chiral center was introduced in the first step, thanks to the lipase reaction. Although compound 51 is obtained in a long sequence (analogous to the above work), the total yield is 1.25%, which gives an average yield of 88% for a step.
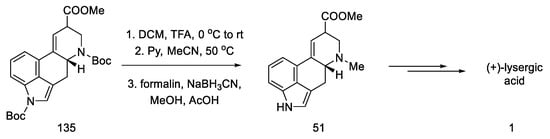
Strategy III. Another synthetic attempt to obtain lysergic acid was undertaken by Fukuyama’s team in 2013 [87]. The acid could be prepared from units with stereogenic centers in allylic positions (136, 137), followed by a D-ring closing metathesis reaction, and finally a Heck reaction to close the C-ring. Synthesis was started with what is known in the literature as crotoylooxazolidinone 138 (Scheme 54) [88].
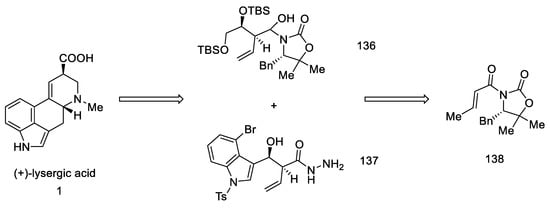
Scheme 54. Retrosynthetic analysis leading to substrates in Fukuyama strategy III.
Compound 138 was prepared, which was in fact an aldehyde masked by oxazolidine. An Evans aldol reaction was used to obtain an amide with a terminal olefin 139. After protection of the -OH groups with TBS, reduction of DIBAL-H was performed to give the hemiaminal 136 (Scheme 55).
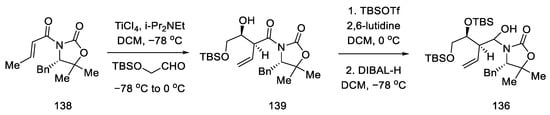
Scheme 55. Preparation of the hemiaminal 136.
The preparation of the second starting material was carried out using the same compound 138. The Evans reaction was performed again—taking advantage of the fact that this aldol reaction allows stereochemical control at the allylic position—using indole aldehyde, since it is known in the literature. Thereafter, treatment with hydrazine gave hydrazide 141, which was converted to the acyl azide. Curtius rearrangement of the azide under acidic conditions gave amine which, upon reduction with triethylsilane, gave allylamine 142 (Scheme 56).
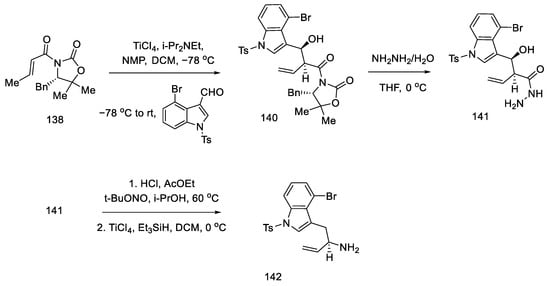
Scheme 56. Preparation of allylamine 142.
Having both compounds—allylamine 142 and the hemiaminal 136—reductive alkylation was performed. The amine was then protected with -Boc, and the D ring was closed using a ring-closing double-bond metathesis reaction. The second-generation Grubbs catalyst was used in this reaction [89], which made it possible to perform cyclization to compound 144, with high efficiency (Scheme 57).
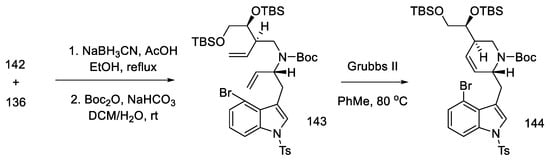
Scheme 57. Reaction of allylamine 142 and the hemiaminal 136, along with reduction by Grubbs catalyst II.
Closing the D-ring using the Heck method yielded tetracyclic product 145, with an ergoline skeleton. Subsequently, the manipulation of protecting groups and functional groups gave ester 51 and then (+)-lysergic acid, analogous to strategies I and II (Scheme 58). Two successive cyclization reactions were the key steps in this procedure.
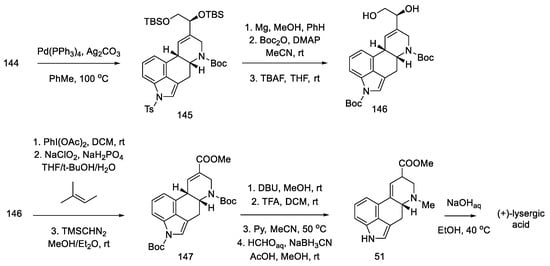
Scheme 58. Preparation of lysergic acid in Fukuyama strategy III.
Strategy IV. The Fukuyama team’s recent work deviates from the idea of using the Heck reaction and instead proposes the formation of a C–C bond between C10 and C11 by aziridine ring cleavage [90]. The reaction begins with the preparation of the substrate—an indoline derivative [91]. Studies have shown that indole undergoes side reactions as a result of knitting the electron-rich 3 position in the bicyclic structure. The choice of the chiral substituent—(3R, 5R, 10R)-trans-aziridine—allowed for the performance of diastereoselective reactions.
The synthesis of the allyl indoline derivative started with 2-bromophenylacetic acid 148. Installation of the chiral auxiliary, followed by allylation and reduction of LiAlH4, gave the alcohol 149. The Mitsunobu reaction was then performed followed by deprotection of the Boc group. The closing of the five-membered cycle of indoline 151 was due to the amination reaction in the presence of copper salts (Scheme 59).
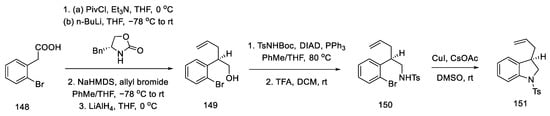
Scheme 59. Beginning of the synthesis according to Fukuyama strategy IV.
Terminal olefin 151 was oxidized to carboxylic acid 152 by treatment with the Jones reagent. The chiral benzyloxazolidin-2-one was reconnected, and a diastereoselective aldol reaction was performed by reacting acyloxazolidinone 153 and an alkyne aldehyde in the presence of a titanium chloride (Scheme 60).
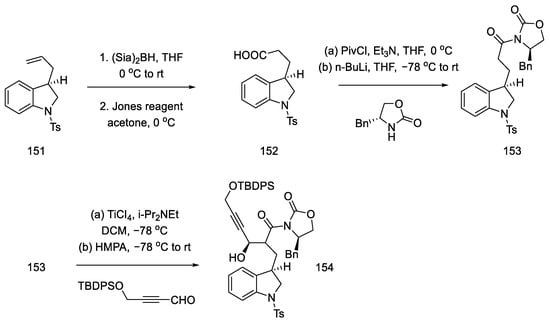
Scheme 60. Preparation of alkyne 154.
The obtained compound 154 was converted to hydrazide using hydrazine and then reacted with nitrite. The formed acyl azide was used for Curtius rearrangement, analogous to the sequence of reactions described in the previous publication, to give oxazolidinone 155 [87]. The necessary stereogenic centers were, thus, incorporated into the compound. In the next step, a nosyl group was inserted in place of the oxazolidinone, which, upon the addition of lithium hydroxide, gave hydroxy-p-Ns-amide 156. The Mitsunobu reaction followed, and trans-aziridine 157 was obtained (Scheme 61).
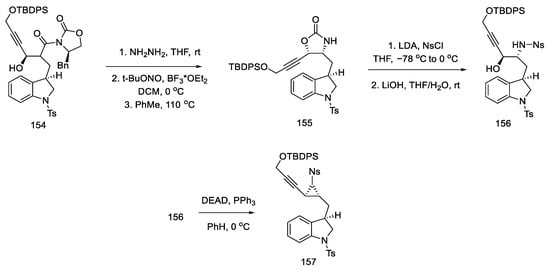
Scheme 61. Synthesis of aziridine 157 in the Mitsunobu reaction.
Upon treatment of aziridine 157 with Lewis acid, the three-membered ring was cleaved stereospecifically. A tricyclic compound containing ABC rings of the ergoline system was obtained. The structure 158 was introduced with an organotin substituent. Upon its use, the D ring was closed by an intramolecular Mitsunobu reaction. The amine was released from the -Ns protecting group with thioglycol acid, followed by methylation with formalin and sodium cyanoborohydride, to give the tetracyclic compound 160 (Scheme 62).
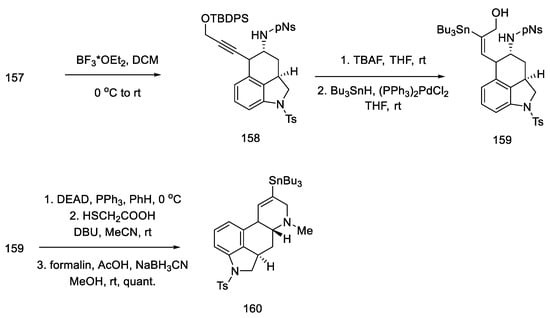
Scheme 62. Closing the D-ring in Fukuyama strategy IV.
After manipulation with protecting groups, indoline 160 was oxidized to indole with (PhSeO)2O. The organotin substituent was replaced with an alkenyl iodide, followed by the incorporation of CO carbon monoxide in the presence of palladium, to obtain an epimeric mixture of esters 162. These esters can be converted to lysergic acid by the methods presented in the previous publications by the Fukuyama group or by the method proposed in this publication by treatment with TFA, decarboxylation by heating with pyridine, and alkaline hydrolysis (Scheme 63).
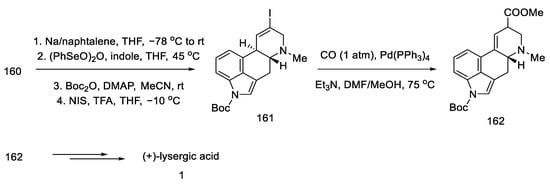
Scheme 63. Preparation of lysergic acid in Fukuyama strategy IV.
Many years of research by the Fukuyama group have led to a number of solutions to the problem of total synthesis of (+)-lysergic acid. All methods are complex syntheses, focused on the gradual preparation of substrates for the C-ring closure reaction, usually the Heck reaction.
2.4.3. Liu and Jia Research
Strategy I. The work of Liu and Jia, from 2011, shows a method of obtaining (+)-lysergic acid in which the starting material is the (R)-derivative of indole 164, as a result of the D ring closing reaction in the double-bond metathesis reaction, followed by the intramolecular Heck reaction [92]. This is a very similar procedure to the one proposed by Fukuyama, but much shorter [85]. The starting indole derivative can be obtained using D-glutamic acid 163 [91].
The synthesis begins with the methylation of the amine with methyl iodide, followed by reduction with borohydride and Swern oxidation. The obtained aldehyde 166 can be used for the Wittig reaction (Scheme 64).
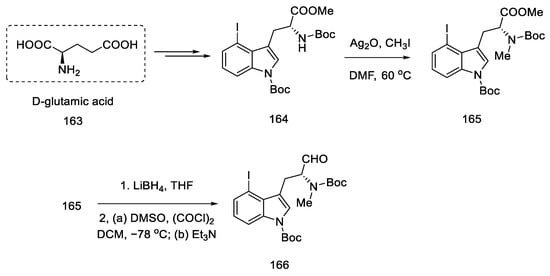
Scheme 64. Beginning of the synthesis of lysergic acid from D-glutamic acid 163.
Enol ether 167 was obtained by treatment with methoxymethylene ylide. Successive reactions gave diene 169, which was a substrate for the D-ring and then C-ring cyclization reaction (Scheme 65).
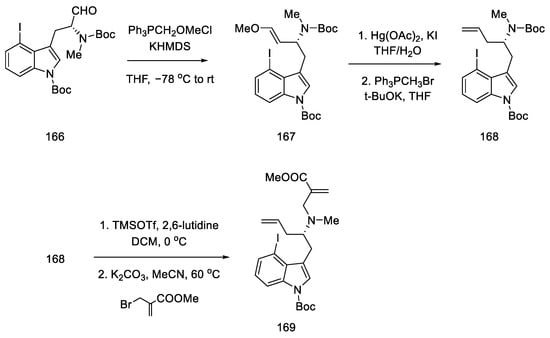
Scheme 65. Introducing of two terminal double bonds into molecule 166 in strategy I.
Compound 169 was treated with a second-generation Grubbs catalyst in the presence of p-TsOH acid [89]. It was tested that, in addition to the presence of a Lewis acid, the choice of solvent (toluene) played a crucial role in the success of the reaction.
Following the synthesis of tricyclic 170, various C-ring closing pathways were investigated. The γ arylation reaction was unsuccessful, so the Heck reaction was tested. After finding the optimized conditions, the reaction was performed in the presence of two equivalents of silver carbonate in hot MeCN to give compound 171 (Scheme 66).
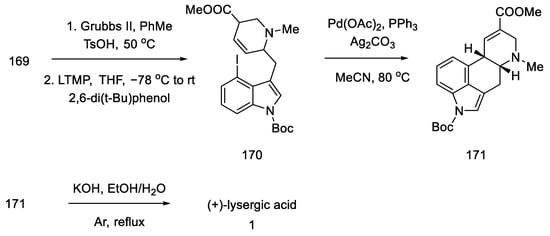
Scheme 66. Method of obtaining lysergic acid proposed by Liu and Jia.
Finally, the Boc group was deprotected, and ester hydrolysis accompanied by double-bond isomerization was performed to obtain (+)-lysergic acid.
Strategy II. The subsequent publication by the Jia team presents an optimized version of strategy I and compares two total syntheses to each other; one is described as strategy I in the above work, plus there is a synthesis in which key stages were swapped (strategy II) [93]. The goal was to further simplify the synthetic path by reducing the effort to protect functional groups. By using 3-chloro-2-iodoaniline, it was possible to simplify the indole coupling to give a compound that could be directly converted into the Heck reaction. The reaction started with simple substrates including chiral (R)-sulfonamide 172.
The first step was to construct a diene that could then be converted to a tetrahydropyridine ring by a metathesis reaction. For this purpose, the aldehyde 173, (R)-N-tert-butanesulfinamide 172, and butenyl bromide were reacted, and the chiral allylamine 174 was obtained in the presence of indium and titanium (dr = 7:1) [94]. In the next step, the reaction with bromomethylacrylate gave diene isomers 175, which were separated by column chromatography (Scheme 67).
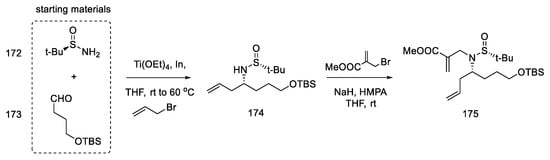
Scheme 67. Introducing two terminal double bonds into the molecule 173 in strategy II.
The metathesis reaction gave the cyclic product 176 by the action of the Grubbs II catalyst in boiling methylene chloride [89]. The -TMS group was deprotected, and the aldehyde 177 was obtained by the Dess–Martin reaction. The obtained product is a fragment of the target structure with the D-ring. The next step was the coupling of the 3-chloro-2-iodoaniline fragment with an aldehyde 177 to synthesize the indole derivative 178 (Scheme 68).
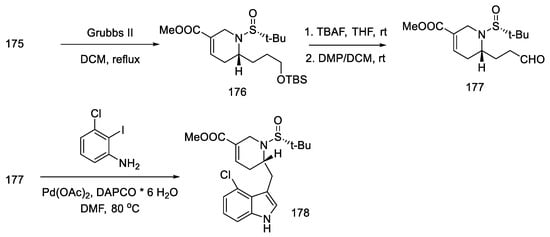
Scheme 68. Synthesis of the ABD tricyclic system in Liu and Jia’s strategy.
Further synthesis relied on manipulating the functional groups to obtain substrate 180 for the intramolecular Heck reaction. To this end, the nitrogen atom in indole was protected with the Boc group, then the tert-butanesulfinyl group was removed, and methyl was introduced onto the nitrogen of the D-ring. Double bond isomerization was performed. Optimization of the Heck reaction was performed. Various reaction conditions were tested (palladium sources, ligands, bases, and solvents). The best results were achieved using 0.5 equivalents of Pd2(dba)3CHCl3, 1.0 equivalents of P(t-Bu)3HBF4, and 3.0 equivalents of Cy2MeN in dioxane at 100 °C. Compound 171, a precursor to lysergic acid, was obtained (Scheme 69). The procedure for obtaining acid therefrom is described in the above works [92].
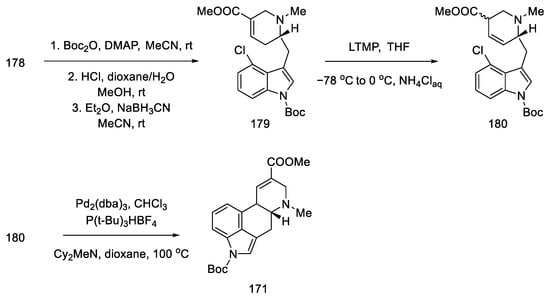
Scheme 69. Preparation of lysergic acid precursor 171.
In two papers presented by the Jia group, two strategies for the preparation of enantioselective acid synthesis of (+)-lysergic acid were described. Both involved metathesis to the D-ring, synthesis of the indole derivative (AB-ring), and the closure of the C-ring by the intramolecular Heck reaction. In strategy I, lysergic acid is obtained in 20 steps. Strategy II allowed for greater convergence of the method and simplified the procedure to just 12 steps.
2.5. Other Chemical Methods of Obtaining Lysergic Acid
2.5.1. Oppolzer Group Research
The Oppolzer group presented the synthesis of (±) lysergic acid, where the C and D rings of the ergoline system were formed through an intramolecular Diels–Alder reaction [84]. This is a convergent reaction using protected 4-hydroxymethylindole as a substrate. The synthetic strategy was based on the preparation of an indole derivative substituted with a diene fragment, then connection of the dienophilic system, followed by cyclization (181 to 182, Scheme 70) and isomerization of the double bond to the lysergic acid.
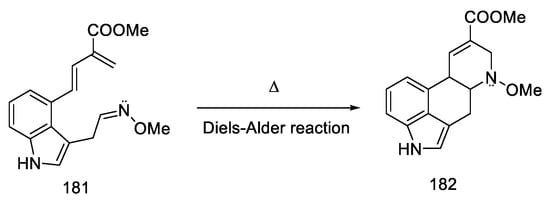
Scheme 70. C and D rings closing strategy in the Diels–Alder reaction.
The synthesis started with the conversion of indole-alcohol 183 to the corresponding bromide with carbon tetrabromide in the Appel reaction. Then the obtained product was treated with phosphine to obtain phosphonium bromide 184 [95].
Difficulties were encountered with introduction of the 2-carbomethoxydiene system into the indole derivative. For this purpose, this moiety was masked with the norbornene fragment. The synthesis of the norbornene derivative 185 was performed using conditions from the literature; formylation of methylbicyclo[2.2.1]hept-5-enyl-2-carboxylate with methyl formate in the presence of LDA gave the expected product 185 [30]. Adding compound 185 into the phosphine salt 184 via the Wittig reaction allowed for obtaining a masked diene moiety in product 186. Then, by nucleophilic addition to nitro compound 187, a fragment of an electron-poor dienophile was attached to the structure (Scheme 71). This reaction was carried out in two ways—using Michael addition to nitroethylene (24% yield) or in a two-step synthesis—by reacting a Mannich reaction with formaldehyde, followed by adding nitromethane in the presence of acetylenedicarboxylate (48%) [96].
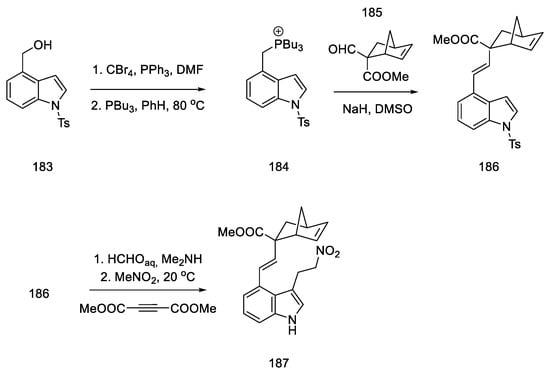
Scheme 71. Introduction of a masked diene system into the molecule 184.
Nitro compound 187 was converted to the oxime 188. The diene moiety was then revealed by retro Diels–Alder thermal reaction, yielding key intermediate 181 proposed by Oppolzer. The intramolecular reaction required keeping the concentration of the intermediate very low throughout the reaction. The dienophile/diene system generated in situ, thus, underwent a Diels–Alder reaction to give the tetracyclic 182 system, as a mixture of diastereomers.
The formal synthesis of lysergic acid was completed by replacing N-methoxy group with methyl group using methyl fluorosulfonate, followed by hydrogenolysis and hydrolysis (Scheme 72). NMR analysis confirmed the identity of the obtained product [57].
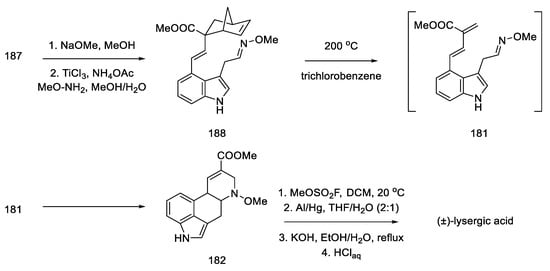
Scheme 72. Preparation of lysergic acid by the Oppolzer procedure.
2.5.2. Research Carried out by Inuki, Iwata. and Ohno
The group led by Ohno was investigating the preparation of heterocyclic compounds by reaction of allenes. These methods were used to synthesize lysergic acid via two synthetic pathways. The key reactions in these pathways were the preparation of the cumulative double bond system giving the allene, followed by the domino reaction of the palladium-catalyzed cyclization of the allene to form a new stereogenic center. This is due to the transfer of the axial chirality of the C=C=C system to the asymmetric carbon [97][98][99].
Strategy I. The synthesis of lysergic acid, lysergol, and isolysergol was carried out. The synthetic strategy was based on the synthesis of allenes by Claisen rearrangement of the corresponding enol ether, followed by cyclization of the CD rings via a palladium-catalyzed domino reaction [97].
The preparation of allenes was proposed by starting the synthesis of 4-bromoindole 61. Reactions from the literature can afford the protected bromomethylindole 189. Using n-butyllithium, a coupling reaction was performed substituting the indole with acetylenecarbaldehyde, with a source that was thioacetal 190. Compound 190 was obtained in 96% yield (Scheme 73).
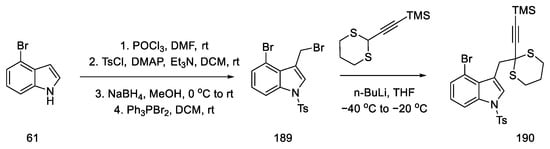
Scheme 73. Synthesis of thioacetal 190 from bromoindole.
Then, by hydrolysis of the thioacetal 190, reduction with sodium borohydride, removal of the TMS protecting group, and addition of methyl propiolate, the enol ether 191 was obtained. In the next reaction, the reduction and protection of the alcohol formed with the TIPS-protected group 192 was carried out (Scheme 74). The compound prepared in this way was a substrate for the Claisen rearrangement reaction, leading to the allene 193.
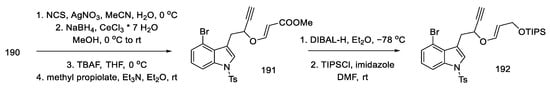
Scheme 74. Enolate ether 192 synthesis.
Optimizing the conditions led the authors to a procedure that used microwave radiation. A diastereomeric mixture of products 193 was obtained, which was used without further purification. The Mitsunobu reaction with NsNH2, analogous with TsNHFmoc, gave N-nosyl and N-tosyl derivatives 194 (Scheme 75). Better results of the following reactions have been achieved by using a tosyl group.
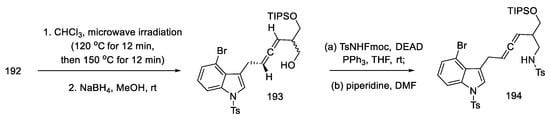
Scheme 75. Obtaining allene in strategy I.
The preparation of the tetracyclic ergoline structure was accomplished by palladium-catalyzed domino cyclization. The best conditions were selected by testing different ligands and bases. The best yield and diastereomeric excess (dr = 83:17 in favor of the desired diastereomer) was obtained with 5% mol Pd(PPh3)4 and 3 eq of potassium carbonate in hot DMF.
Tosyl-protected compound 195 was a lysergic acid precursor. Cleavage of the TIPS group, oxidation of the resulting alcohol in the Dess–Martin procedure, and esterification gave the product 196. Then, the NH groups were deprotected, and the ester 51 used in the synthesis of lysergic acid was obtained (Scheme 76), which can be converted into the desired, optically pure acid by alkaline and acid hydrolysis.
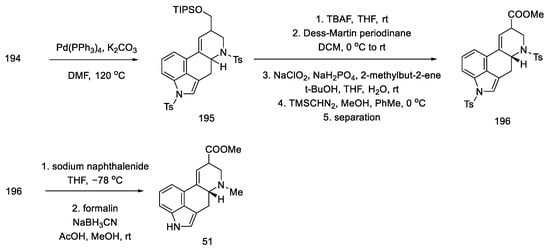
Scheme 76. Preparation of lysergic acid ester 51 proposed by Inuki, Iwata, and Ohno.
Strategy II. The allene was obtained by the reductive coupling reaction of aziridine palladium and the creation of the C-C bond between the indole derivative and oxazinane, followed by the Myers reaction [99].
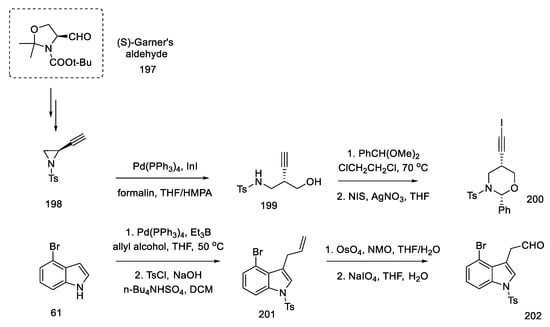
First, the materials for the coupling were prepared. For this purpose, bromoindole aldehyde 202 was prepared from bromoindole 61, using the coupling reaction of allyl alcohol according to the procedure in the literature [100], protection of the nitrogen atom in the indole, and then oxidative cleavage of olefin 201 to aldehyde 202. The second substrate was the chiral oxazinane derivative 200. It was prepared from optically pure aziridine 198 (97% ee), which, in turn, is obtainable from (S)-Garner aldehyde 197 in a four-step synthesis [101]. By treating aziridine 198 with a catalytic amount of a palladium compound with InI in a mixture of THF/HMPA and formalin, the amino alcohol 199 was obtained, which could be easily changed into the oxazinane derivative 200 (Scheme 77). Both iodinated and free terminal alkyne enable the coupling reaction to be carried out; however, by introducing iodine, the efficiency and selectivity of the coupling process increase.
Scheme 77. Synthesis of materials for the coupling in strategy II.
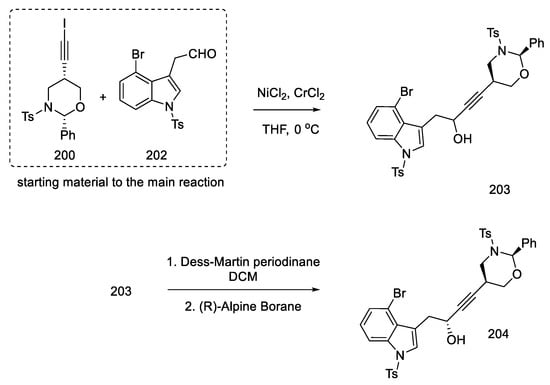
With the prepared substrates 200 and 202 in hand, the process of their combination was started. The coupling reaction of these fragments was carried out using nickel chloride and chromium chloride. A racemic mixture of alcohol 203 was obtained. Dess–Martin oxidation followed by reduction with (R)-Alpine Borane made it possible to obtain the desired asymmetric product 204 from the racemic alcohol (dr = 95:5) (Scheme 78).
Scheme 78. Reaction leads to an asymmetric alcohol 204.
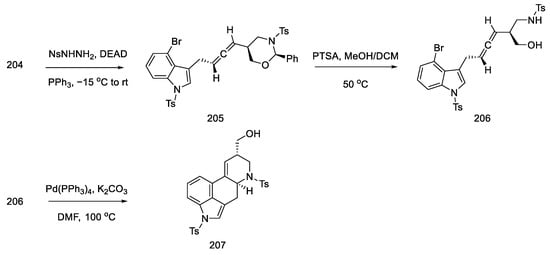
Alcohol 204 was stereoselectively converted to the allene 205 by the Myers method [102], using nitrobenzenesulfonyl hydrazine under Mitsunobu conditions. Then the oxazinane was cleaved with PTSA. The product 206 was obtained with a diastereomeric excess (94:6), but the mixture was not separated. A domino cyclization reaction through palladium compounds was performed under optimized conditions (5 mol% Pd(PPh3)4, K2CO3, and DMF at 100 °C). The analogous reaction for the second epimeric product was also checked using reduction with (S)-borane. The cyclization reaction from the allene gave the corresponding diastereoisomer 207 with high selectivity. This compound is the key intermediate in the approach used by the Ohno group (Scheme 79).
Scheme 79. Obtaining allene in strategy II.
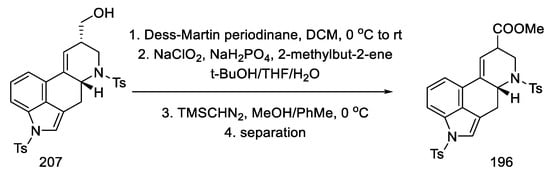
The primary alcohol 207 was oxidized with a Dess–Martin reagent and chlorite. The esterification was then carried out to the known racemic methyl ester, of which both amine functions were protected with tosyl (196), which could be transformed into lysergic acid (Scheme 80) [63][69][97].
Scheme 80. Obtaining intermediate 196 in strategy II.
The above publication describes methods for the synthesis of other ergot alkaloids, lysergol and isolysergol. The total synthesis of lysergic acid consists of 19 steps, and the overall yield is 1.1%. The starting material was Garner’s aldehyde. The innovation of this synthetic strategy involves the construction of the D ring as a result of catalytic cyclization of the allene. Further work by Ohno’s team boiled down to new methods of obtaining the above allene, which can then be easily transformed into the intermediate 51.
Strategy III. Strategy III of obtaining lysergic acid—specifically of intermediate 51—was based on similar assumptions as strategy II. The bromoindole derivative 61 and the corresponding alkyne were prepared, and the coupling reaction was performed. The resulting alkyne was converted to an allene, which could be converted to the corresponding intermediate. The new idea was to obtain a chiral alcohol, not by an asymmetric oxidation reaction, but by a regioselective epoxide opening sequence. For this purpose, the reactants were modified—the synthesis started with bromoindole and the corresponding sulfonamide terminal (S)-alkyne, as known in the literature [98].
Commercially available 4-bromoindole 61 was formylated by the Vilsmeier–Haack reaction [103], followed by treatment of resulting aldehyde 208 with CBr4, which gave dibromoolefin 209. Using the Uenishi method [104], followed by the Sonogashira coupling of the known chiral alkyne, which can be prepared from (S)-Garner aldehyde 197 (analogous to strategy I), a compound was obtained containing a triple and a double bond at adjacent carbon atoms—enyne 210 (Scheme 81).
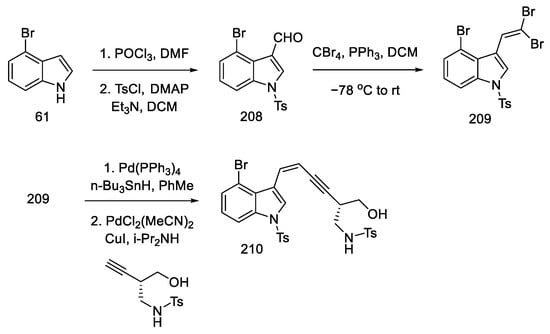
Scheme 81. Synthesis of enyne 210 from bromoindole.
The epoxidation of enyne 210 was a challenging step. Various epoxidizing agents were tested, but neither the use of the Shi catalyst nor the m-CPBA acid allowed for obtaining the epoxide 211 in a satisfactory yield. Only the optimization of the conditions (presence of n Bu4NHSO4, oxone) allowed the yield of the product to be over 70%, with a diastereomeric ratio of 82:18. The OH group was protected with TIPSOT, and then chiral alcohol 212 was obtained by ring opening with Lewis acid (ZnCl2 or Zn(OTf)2), retaining the excess diastereoisomer. Alcohol 212 was converted (analogycal to strategy II) in allene 206 using a modified Myers reaction via nitrobenzenesulfonyl hydrazine (Scheme 82) [105].
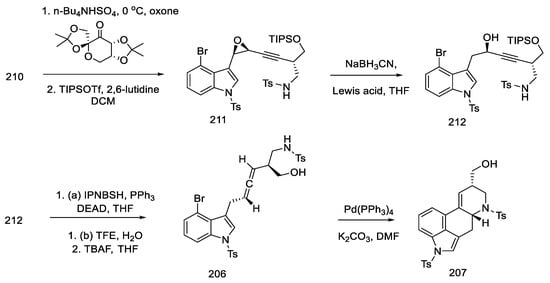
Scheme 82. Obtaining allene in strategy III.
In summary, strategy III used the following reactions: obtaining a bromoolefin, a derivative of indole 209; then, via the Uenishi reaction and Sonogashira coupling to a terminal alkyne, the enyne, thus obtained, was oxidized by an asymmetric epoxidation reaction, and the alkene oxide was opened by regioselective reduction to the chiral alcohol. This alcohol, with a carbon–carbon triple bond in its structure, was converted into an allene in a modified Myers reaction, which, analogically to strategy I and II, can be converted into intermediate 51, and, further, into (+)-lysergic acid.
2.6. Attempts of Obtaining Lysergic Acid by Other Methods
2.6.1. Padwa Group Research
In 1995, the Albert Padwa research group developed a new synthetic pathway that could eventually lead to lysergic acid [106].
First, it was shown that the intramolecular dipolar cycloaddition of isomuenchnone by an olefin or an alkyne leads to the quinoline structure—the C and D ring of ergot alkaloids. Subsequently, a 12-step synthesis was carried out, starting from the known tricyclic olefin 215, through diazoimide 214, to lysergic acid derivative 213 (Scheme 83). Unfortunately, the double-bond isomerization, which would lead to a lysergic acid structure, could not be carried out.
Scheme 83. Retrosynthetic analysis proposed by Padwa.
The synthesis began with the known protected tetrahydrobenzoindole 215. Ozonolysis was performed followed by reduction with sodium borohydride. A diol was obtained, which was selectively oxidized, followed by a Wittig reaction, to give olefin 216. In the next three steps, amide 217 was obtained from the olefin using the Jones reagent, carbonyldiimidazole, and methylamine. Diazoimide 214 was prepared by the action of acid chloride and azide. This compound was used to study the cyclization reaction using the Rh(II) catalyst (Scheme 84) [107].
Scheme 84. Beginning of the synthesis proposed by Padwa.
The cyclization conditions have been optimized by selecting the best catalyst. The best selectivity was achieved using perfluorobutyrate (pfb) as the rhodium catalyst ligand.
The conversion of the oxygen bridge containing product 215 to the corresponding enamide 216 was performed using boron fluoride as a Lewis acid. The enamide, thus obtained, was treated with thiocarbonate in benzene by performing the Barton–McCombie reaction [108]. Chiral ester 213 was obtained in the form of a mixture of diastereoisomers. Unfortunately, all attempts to isomerize the double bond have been unsuccessful (Scheme 85).
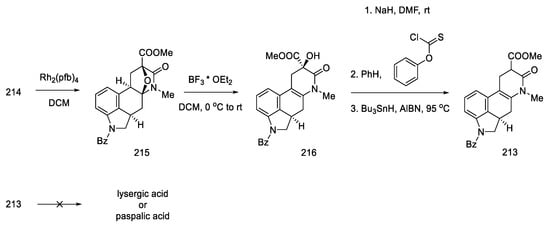
Scheme 85. Continuation of the synthesis proposed by Padwa.
In conclusion, the total synthesis of ergot alkaloids—lysergic acid and paspalic acid—could not be completed. At the same time, the method proposed by Padwa allows for the closure of the C and D rings to the ergoline structure, and further research may lead to the achievement of the assumed goal.
2.6.2. Parsons Group Research
One year after the Padwa research, an article by the Parsons group was published, which presented the results of new methods of performing tandem reactions for the ring closure in natural compounds and their analogues [109]. This publication summarizes the team’s achievements, including the possibility of closing three- and four-membered systems leading to ergoline, as presented previously [110]. There are also studies conducted by Ȍzlȕ and Cladingboel, who attempted to synthesize lysergic acid [111]. In these studies, free radical reactions obtained by homolytic cleavage of a carbon–bromine bond with a hydride were used. Radical reactions allow single or double (tandem) cyclization reaction to take place.
The starting material was 2-bromo-3-nitrobenzoic acid 217. As a result of the multistage synthesis, the aldehyde 218 was obtained, which was then reacted with amines to give compounds 219 and 221.
Cladingboel proposed a radical cyclization reaction using MPM-protected compound 219. Tandem cyclization, performed with tributyltin hydride and AIBN, resulted in reduced to a five-membered structure of the D-ring 220.
Continuing this work, Ȍzlȕ used methyl 2-(methylamine)acrylate to introduction an ester group in compound 221. Operating at elevated temperature, cyclization of the six-membered D-ring was performed; however, the radical reaction also led not to lysergic acid but to its saturated derivative 223 (Scheme 86).
Scheme 86. Parsons group synthetic strategies.
2.7. Biosynthetic Pathways of Lysergic Acid
The pioneer of the topic of ergot alkaloids biosynthesis was Floss, who, in 1976, presented a set of information on the synthesis processes in biological systems. Since then, many processes have been clarified and the related knowledge has been expanded [112].
A study by Shu-Ming Li’s team, published in 2014, outlines the biochemical pathways leading to ergot alkaloids, including lysergic acid [9]. The study collected information on intermediates, the enzymes used as biocatalysts, and the genes that allowed specific reactions to take place. Not all the mechanisms have been confirmed, but the identification of intermediates allows for understanding the most likely reaction mechanisms. Similarly, Mukherjee and Joydeep, in 2000, put together strategies to chemically and biochemically synthesize ergot alkaloids [113].
Lysergic acid biosynthesis begins with L-tryptophan. In the first step, C4-prenylation was carried out with the use of an enzyme isolated from Claviceps sp., which is encoded by the dmaW gene [114]. For this purpose, the biochemical reaction takes place with dimethylallyl diphosphate as a prenyl donor, in the presence of a biocatalyst—DMATS synthase prenyltransferase [115]. The mechanism of this reaction, proposed by Metzger, resulted from the X-ray structure of the active complex. It includes three stages: formation of the dimethylallyl cation, nucleophilic attack of the indole nucleus to the cation, and deprotonation [116]. The second step in biosynthesis is N-methylation of allyl-tryptophan 224 to the L-abrine derivative 225, catalyzed by EasF methyltransferase in the presence of (S)-adenosylmethionine SAM (Scheme 87) [117].
Scheme 87. The first stages of lysergic acid biosynthesis.
The mechanism of conversion of compound 225 to 228 is not confirmed. It is known that this reaction requires decarboxylation and oxidation steps using at least two enzymes—FAD-dependent oxidoreductase (FgaOx1) and FgaCat catalase, in which easE and EasC genes play a key role. Presumably, this process occurs through the steps of: desaturation of the C8–C9 bond by hydroxylation of the benzyl carbon, deprotonation of the C17 carbon, oxidation of the C7–C8 bond, and decarboxylation of the epoxide 227 intermediate (Scheme 88) [118][119][120][121].
Scheme 88. Biomechanism proposed by Metzger.
The next step in the synthesis leads to the aldehyde 229. It is obtained by a short-chain dehydrogenase/reductase-catalyzed reaction SDR. Wallwey used FgaDH for this purpose in the presence of NAD+ [122]. Aldehyde 229 is an important intermediate, from which compounds such as festuclavine, pyroclavine, and agroclavine can be obtained through the biochemical pathway of fungi. The four-ring agroclavine 231 can be obtained from Claviceps purpurea by treating the aldehyde of the enzyme from E. festucae var. lolii or EasG, as a result of which an adduct with reduced glutathione is transformed into the desired intermediate (Scheme 89) [123][124].
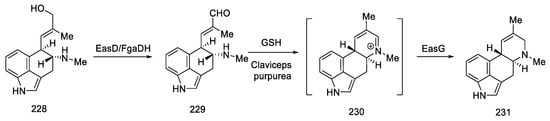
Scheme 89. Enzymatic cyclization of the D-ring.
The biosynthesis of lysergic acid from agroclavine 231 remains unexplained. Among the intermediates leading to the final product, elymoclavine 232 and paspalic acid 233 can be distinguished. It is postulated that cytochrome P-450 monooxygenase, or rather its gene—cloA, is an essential enzyme for subsequent transitions. Elimoclavin can be obtained by the two-electron oxidation of agroclavin [125]. Then the application of four-electron reduction leads to paspalic acid 233 [125][126]. Paspalic acid can be converted to lysergic acid either by the action of isomerase or by spontaneous rearrangements (Scheme 90) [112].
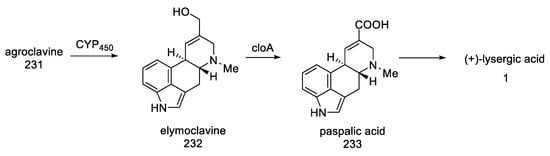
Scheme 90. Preparation of lysergic acid from agroclavine.
The genes for making ergot alkaloids are grouped together in the producer’s genomes. By genetic modification of the host, the amount and type of ergot alkaloids produced can be varied [127].
At this point, it is worth mentioning the publication “Recent progress in ergot alkaloid research” by Ping Zhu [128]. In addition to proposing the mechanisms of action of some ergot alkaloids, they presented the characteristics of the key genes and gene clusters involved in the biosynthetic process leading, inter alia, to lysergic acid. In particular, the focus was on genetically modifying the strains, so that the main acid and its derivatives can be produced through a biological engineering strategy (Scheme 91).
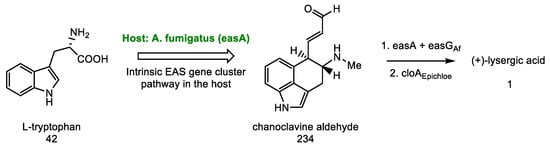
Scheme 91. Lysergic acid biosynthesis strategy proposed by Zhu.
An exemplary strategy is to start the biosynthesis with L-tryptophan 42 and DMAPP. Next, in the modified host organism, there are transformations from chanoclavine to aldehyde 234, and then through agroclavine to lysergic acid [129].
As mentioned at the beginning, the annual production of lysergic acid is over 10 tons per year. Chemical total syntheses are characterized by low total yields; therefore, biotechnological methods are implemented. In 2022, an analogous biosynthetic pathway (starting with tryptophan), but using a modified yeast chassis of the DDM33B strain, was used to investigate the amount of acid that could be obtained from a biotechnological batch culture. The scale-up studies allowed for estimating that 52 g of lysergic acid can be obtained per year using the 1000 L bioreactor [130].
This entry is adapted from the peer-reviewed paper 10.3390/molecules27217322
References
- Ochodzki, P.; National Research Institute. Zapobieganie Zagrożeniom Związanym z Występowaniem w Ziarnie Zbóż Alkaloidów Pasożytniczego Grzyba Buławinki Czerwonej (Sporysz)—Prevention of Risks Associated with the Presence of Alkaloids in the Cereal Grain Parasitic Fungus of Ergot; Expertise: Radzików, Poland, 2015.
- Schiff, P.L. Ergot and Its Alkaloids. Am. J. Pharm. Educ. 2006, 70, 98.
- Smakosz, A.; Kurzyna, W.; Rudko, M.; Dąsal, M. The Usage of Ergot (Claviceps Purpurea (Fr.) Tul.) in Obstetrics and Gynecology: A Historical Perspective. Toxins 2021, 13, 492.
- Krska, R.; Crews, C. Significance, Chemistry and Determination of Ergot Alkaloids: A Review. Food Addit. Contam. Part A 2008, 25, 722–731.
- Berde, B.; Schild, H.O. (Eds.) Ergot Alkaloids and Related Compounds; Springer: Berlin/Heidelberg, Germany, 1978.
- Stoll, A.; Petrzilka, T.; Rutschmann, J.; Hofmann, A.; Günthard, H.H. Über Die Stereochemie Der Lysergsäuren Und Der Dihydro-Lysergsäuren. 37. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1954, 37, 2039–2057.
- Hofmann, A.; Ott, H.; Griot, R.; Stadler, P.A.; Frey, A.J. Die Synthese Und Stereochemie Des Ergotamins. (58. Mitteilung Über Mutterkornalkaloide). Helv. Chim. Acta 1963, 46, 2306–2328.
- Flieger, M.; Wurst, M.; Shelby, R. Ergot Alkaloids—Sources, Structures and Analytical Methods. Folia Microbiol. 1997, 42, 3–30.
- Gerhards, N.; Neubauer, L.; Tudzynski, P.; Li, S.-M. Biosynthetic Pathways of Ergot Alkaloids. Toxins 2014, 6, 3281–3295.
- Matławska, I. Farmakognozja; UM: Poznań, Poland, 2008.
- Seeman, P. Historical Overview: Introduction to the Dopamine Receptors. In The Dopamine Receptors; Humana Press: Totowa, NJ, USA, 2010; pp. 1–21.
- Pucadyil, T.J.; Kalipatnapu, S.; Chattopadhyay, A. The Serotonin1A A Receptor: A Representative Member of the Serotonin Receptor Family. Cell. Mol. Neurobiol. 2005, 25, 553–580.
- Halberstadt, A.L.; Geyer, M.A. Multiple Receptors Contribute to the Behavioral Effects of Indoleamine Hallucinogens. Neuropharmacology 2011, 61, 364–381.
- Nichols, D.E. Psychedelics. Pharmacol. Rev. 2016, 68, 264–355.
- Watts, V.J.; Mailman, R.B.; Lawler, C.P.; Neve, K.A.; Nichols, D.E. LSD and Structural Analogs: Pharmacological Evaluation at D1 Dopamine Receptors. Psychopharmacology 1995, 118, 401–409.
- Bijoch, Ł.; Pękała, M.; Beroun, A. Molekularne Podstawy Działania Wybranych Substancji Psychoaktywnych. Postepy Biochem. 2021, 67, 141–156.
- Fuentes, J.J.; Fonseca, F.; Elices, M.; Farré, M.; Torrens, M. Therapeutic Use of LSD in Psychiatry: A Systematic Review of Randomized-Controlled Clinical Trials. Front. Psychiatry 2020, 10, 943.
- Smith, D.E.; Raswyck, G.E.; Dickerson Davidson, L. From Hofmann to the Haight Ashbury, and into the Future: The Past and Potential of Lysergic Acid Diethlyamide. J. Psychoact. Drugs 2014, 46, 3–10.
- Johnson, J.W.; Ellis, M.J.; Piquette, Z.A.; MacNair, C.; Carfrae, L.; Bhando, T.; Ritchie, N.E.; Saliba, P.; Brown, E.D.; Magolan, J. Antibacterial Activity of Metergoline Analogues: Revisiting the Ergot Alkaloid Scaffold for Antibiotic Discovery. ACS Med. Chem. Lett. 2022, 13, 284–291.
- Webster, J. Dopamine Agonist Therapy in Hyperprolactinemia. J. Reprod. Med. 1999, 44, 1105–1110.
- Hamon, M.; Mallat, M.; Herbet, A.; Nelson, D.L.; Audinot, M.; Pichat, L.; Glowinski, J. Metergoline: A New Ligand of Serotonin Receptors in the Rat Brain. J. Neurochem. 1981, 36, 613–626.
- Miller, K.J.; King, A.; Demchyshyn, L.; Niznik, H.; Teitler, M. Agonist Activity of Sumatriptan and Metergoline at the Human 5-HT1Dβ Receptor: Further Evidence for a Role of the 5-HT1D Receptor in the Action of Sumatriptan. Eur. J. Pharmacol. Mol. Pharmacol. 1992, 227, 99–102.
- Hofmann, A. Historical View on Ergot Alkaloids. Pharmacology 1978, 16, 1–11.
- Barger, G.; Carr, F.H. The Alkaloids of Ergot. J. Chem. Soc. Trans. 1907, 91, 337–353.
- Barger, G.; Dale, H.H. Ergotoxine and Some Other Constituents of Ergot. Biochem. J. 1907, 2, 240–299.
- Stoll, A. Zur Kenntnis Der Mutterkornalkaloide. Verh. Schweiz. Nat. Ges. 1918, 101, 190–191.
- Rothlin, E.; Konzett, H.; Cerletti, A. The antagonism of ergot alkaloids towards the inhibitory response of the isolated rabbit intestine to epinephrine and norepinephrine. J. Pharmacol. Exp. Ther. 1954, 112, 185–190.
- Jacobs, W.A.; Craig, L.C. The structure of the ergot alkaloids. J. Am. Chem. Soc. 1935, 57, 383–384.
- Stoll, A.; Hofmann, A.; Troxler, F. Über Die Isomerie von Lysergsäure Und Isolysergsäure. 14. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1949, 32, 506–521.
- Kornfeld, E.C.; Fornefeld, E.J.; Kline, G.B.; Mann, M.J.; Morrison, D.E.; Jones, R.G.; Woodward, R.B. The Total Synthesis of Lysergic Acid. J. Am. Chem. Soc. 1956, 78, 3087–3114.
- Ramage, R.; Armstrong, V.W.; Coulton, S. A New Synthetic Route to (±) Lysergic Acid. Tetrahedron 1981, 37, 157–164.
- Stadler, P.A.; Frey, A.J.; Troxler, F.; Hofmann, A. Selektive Reduktions- Und Oxydationsreaktionen an Lysergsäure- Derivaten. 2.3-Dihydro- Und 12-Hydroxy-Lysergsäureamide. 59. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1964, 47, 756–769.
- Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry. Angew. Chem. Int. Ed. Engl. 1969, 8, 781–853.
- Stoll, A.; Hofmann, A. Die Optisch Aktiven Hydrazide Der Lysergsäure Und Der Isolysergsäure. (4. Mitteilung Über Mutterkornalkaloide). Helv. Chim. Acta 1943, 26, 922–928.
- Woodward, R.B. Private Communication between Woodward and Ramage. Ph.D. Thesis, Harvard, Cambridge, MA, USA, 1952.
- Křen, V.; Cvak, V. Ergot The Genus Claviceps (Medicinal and Aromatic Plants—Industrial Profiles); CRC Press: Boca Raton, FL, USA; Informa UK Limited: London, England, 1999; Volume 6.
- Martínková, L.; Křen, V.; Cvak, L.; Ovesná, M.; Přepechalová, I. Hydrolysis of Lysergamide to Lysergic Acid by Rhodococcus Equi A4. J. Biotechnol. 2000, 84, 63–66.
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1942.
- Blount, B.K.; Robinson, R. CCCCXXXVI.—Strychnine and Brucine. Part XIII. Note on Dihydroindolylpropionic and Dihydroindolylbutyric Acids. J. Chem. Soc. 1931, 3158–3160.
- Uhle, F.C. The Synthesis of 5-Keto-1,3,4,5-TetrahydrobenzIndole. A Synthesis of 4-Substituted Indoles. J. Am. Chem. Soc. 1949, 71, 761–766.
- Johnson, W.S.; Belew, J.S.; Chinn, L.J.; Hunt, R.H. Studies Relating to the Formation and Reactions of Glycidic Esters. J. Am. Chem. Soc. 1953, 75, 4995–5001.
- Price, C.C.; Krishnamurti, I.V. The Acid-Catalyzed Reaction of Formaldehyde with Allyl Cyanide. J. Am. Chem. Soc. 1950, 72, 5334–5335.
- Kleiderer, E.C.; Kornfeld, E.C. Raney nickel as an organic oxidation-reduction catalyst. J. Org. Chem. 1948, 13, 455–458.
- Stoll, A.; Hofmann, A. Racemische Lysergsäure Und Ihre Auflösung in Die Optischen Antipoden.-Zweite, Vorläufige Mitteilung Über Mutterkornalkaloide. Hoppe Seyler’s Z. Für Physiol. Chem. 1937, 250, 7–10.
- Smith, S.; Timmis, G.M. A New Alkaloid of Ergot. Nature 1936, 137, 111.
- Julia, M.; Le Goffic, F.; Igolen, J.; Baillarge, M. Une Nouvelle Synthese de l’acide Lysergique. Tetrahedron Lett. 1969, 10, 1569–1571.
- House, H.O.; Rasmussen, G.H. Stereoselective Synthesis of α-Substituted α,β-Unsaturated Esters. J. Org. Chem. 1961, 26, 4278.
- Jackson, A.G.; Kenner, G.W.; Moore, G.A.; Ramage, R.; Thorpe, W.D. Activation of Carboxylic Acids as Diphenylphosphinic Mixed Anhydrides: Application to Peptide Chemistry. Tetrahedron Lett. 1976, 17, 3627–3630.
- Bu’Lock, J.D.; Harley-Mason, J. 497. Melanin and Its Precursors. Part III. New Syntheses of 5: 6-Dihydroxyindole and Its Derivatives. J. Chem. Soc. 1951, 2248–2252.
- Troxler, F. Beiträge Zur Chemie Der 6-Methy1-8-Ergolen-8-Carbonsäure. 69. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1968, 51, 1372–1381.
- Eschweiler, W. Ersatz von an Stickstoff Gebundenen Wasserstoffatomen Durch Die Methylgruppe Mit Hülfe von Formaldehyd. Ber. Dtsch. Chem. Ges. 1905, 38, 880–882.
- Clarke, H.T.; Gillespie, H.B.; Weisshaus, S.Z. The Action of Formaldehyde on Amines and Amino Acids 1. J. Am. Chem. Soc. 1933, 55, 4571–4587.
- Fehr, T.; Stadler, P.A.; Hofmann, A. Demethylierung Des Lysergsäuregerüstes. 73. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1970, 53, 2197–2201.
- Rebek, J.; Tai, D.F. A New Synthesis of Lysergic Acid. Tetrahedron Lett. 1983, 24, 859–860.
- Rebek, J.; Tai, D.F.; Shue, Y.K. Synthesis of Ergot Alkaloids from Tryptophan. J. Am. Chem. Soc. 1984, 106, 1813–1819.
- Hanessian, S. Selective Hydrolysis of Amide Bonds in Acetamido Deoxy Sugars.-Ethyl Acetamidium Fluoroborates. Tetrahedron Lett. 1967, 8, 1549–1552.
- Smith, S.; Timmis, G.M. 311. The Alkaloids of Ergot. Part VII. IsoErgine and Isolysergic Acids. J. Chem. Soc. 1936, 1440–1444.
- Ninomiya, I.; Kiguchi, T.; Hashimoto, C.; Naito, T. A New Synthesis of (±)-Lysergic Acid. Heterocycles 1982, 19, 2279.
- Ninomiya, I.; Naito, T. Enamide Cyclization and Its Application to the Synthesis of Heterocycles. J. Synth. Org. Chem. Japan 1984, 42, 225–246.
- Nichols, D.E.; Robinson, J.M.; Li, G.S.; Cassady, J.M.; Floss, H.G. An improved synthesis of 1-benzoyl-4-keto-1,2,2a,3,4,5-hexahydrobenz indole. Org. Prep. Proced. Int. 1977, 9, 277–280.
- Ninomiya, I.; Hashimoto, C.; Kiguchi, T.; Naito, T. Photocyclisation of Enamides. Part 24. Total Synthesis of (±)-Isofumigaclavine B and (±)-Lysergic Acid. J. Chem. Soc. Perkin Trans. 1 1985, 941–948.
- Tasker, N.R.; Wipf, P. A Short Synthesis of Ergot Alkaloids and Evaluation of the 5-HT 1/2 Receptor Selectivity of Lysergols and Isolysergols. Org. Lett. 2022, 24, 7255–7259.
- Hendrickson, J.B.; Wang, J. A New Synthesis of Lysergic Acid. Org. Lett. 2004, 6, 3–5.
- Doll, M.K.-H. A Short Synthesis of the 8-Azaergoline Ring System by Intramolecular Tandem Decarboxylation−Cyclization of the Minisci-Type Reaction. J. Org. Chem. 1999, 64, 1372–1374.
- Bekkam, M.; Mo, H.; Nichols, D.E. A Reported “New Synthesis of Lysergic Acid” Yields Only The Derailment Product: Methyl 5-Methoxy-4,5-DihydroindoloQuinoline-9-Carboxylate. Org. Lett. 2012, 14, 296–298.
- Zhao, Y. Reduction of Tertiary Benzamides to Benzaldehydes by an in Situ–Generated Schwartz Reagent (Cp2Zr(H)Cl); Formal Synthesis of Lysergic Acid; Queen’s University Kingston: Kingston, ON, Canada, 2010.
- Points, G.L.; Stout, K.T.; Beaudry, C.M. Regioselective Formation of Substituted Indoles: Formal Synthesis of Lysergic Acid. Chem. Eur. J. 2020, 26, 16655–16658.
- Zhao, Y.; Snieckus, V. A Practical in Situ Generation of the Schwartz Reagent. Reduction of Tertiary Amides to Aldehydes and Hydrozirconation. Org. Lett. 2014, 16, 390–393.
- Moldvai, I.; Temesvári-Major, E.; Incze, M.; Szentirmay, É.; Gács-Baitz, E.; Szántay, C. Enantioefficient Synthesis of α-Ergocryptine: First Direct Synthesis of (+)-Lysergic Acid. J. Org. Chem. 2004, 69, 5993–6000.
- Teranishi, K.; Hayashi, S.; Nakatsuka, S.; Goto, T. A Facile Biomimetic Synthesis of Uhle’s Ketone by the Regioselective Friedel-Crafts Cyclization of Indole-3-Ylpropionyl Chloride. Tetrahedron Lett. 1994, 35, 8173–8176.
- Roth, M.; Dubs, P.; Götschi, E.; Eschenmoser, A. Sulfidkontraktion via Alkylative Kupplung: Eine Methode Zur Darstellung von β-Dicarbonylderivaten. Über Synthetische Methoden, 1. Mitteilung. Helv. Chim. Acta 1971, 54, 710–734.
- Moldvai, I.; Temesvári-Major, E.; Incze, M.; Dörnyei, G.; Szentirmay, É.; Szántay, C. Synthetic Route to Ergot Alkaloids. Helv. Chim. Acta 2005, 88, 1344–1356.
- Rathnayake, U.; Garner, P. Asymmetric Synthesis of Lysergic Acid via an Intramolecular (3+2) Dipolar Cycloaddition/Ring-Expansion Sequence. Org. Lett. 2021, 23, 6756–6759.
- Zuo, Z.; Ma, D. Enantioselective Total Syntheses of Communesins A and B. Angew. Chem. Int. Ed. Engl. 2011, 50, 12008–12011.
- Métro, T.-X.; Appenzeller, J.; Gomez Pardo, D.; Cossy, J. Highly Enantioselective Synthesis of β-Amino Alcohols. Org. Lett. 2006, 8, 3509–3512.
- Jarvis, S.B.D.; Charette, A.B. Synthesis of Enantiopure Substituted Piperidines via an Aziridinium Ring Expansion. Org. Lett. 2011, 13, 3830–3833.
- Oestreich, M. (Ed.) The Mizoroki–Heck Reaction; John Wiley & Sons, Ltd.: Chichester, UK, 2009.
- Cacchi, S.; Giuseppe Ciattini, P.; Morera, E.; Ortar, G. A Concise, Palladium-Catalyzed Approach to (±)-Lysergic Acid. Tetrahedron Lett. 1988, 29, 3117–3120.
- Kurihara, T.; Terada, T.; Yoneda, R. A New Synthesis of (±)-Lysergic Acid. Chem. Pharm. Bull. 1986, 34, 442–443.
- Kurihara, T.; Terada, T.; Harusawa, S.; Yoneda, R. Synthetic Studies of (±)-Lysergic Acid and Related Compounds. Chem. Pharm. Bull. 1987, 35, 4793–4802.
- Busqué, F.; de March, P.; Figueredo, M.; Font, J.; Gallagher, T.; Milán, S. Efficient Synthesis of (S)-3,4-Dihydro-2-Pivaloyloxymethyl-2H-Pyrrole 1-Oxide. Tetrahedron Asymmetry 2002, 13, 437–445.
- Birkofer, L.; Bierwirth, E.; Ritter, A. Decarbobenzoxylierungen Mit Triäthylsilan. Chem. Ber. 1961, 94, 821–824.
- Ghosez, L.; Haveaux, B.; Viehe, H.G. Alkyl and Arylα-Chloro Enamines. Angew. Chem. Int. Ed. Engl. 1969, 8, 454–455.
- Oppolzer, W.; Francotte, E.; Bättig, K. Total Synthesis of (±)-Lysergic Acid by an Intramolecular Imino- Diels—Alder Reaction. Preliminary Communication. Helv. Chim. Acta 1981, 64, 478–481.
- Kurokawa, T.; Isomura, M.; Tokuyama, H.; Fukuyama, T. Synthesis of Lysergic Acid Methyl Ester via the Double Cyclization Strategy. Synlett 2009, 2009, 775–778.
- Ninomiya, I.; Hashimoto, C.; Kiguchi, T.; Naito, T.; Barton, D.H.R.; Lusinchi, X.; Milliet, P. Dehydrogenation with Benzeneseleninic Anhydride in the Total Synthesis of Ergot Alkaloids. J. Chem. Soc. Perkin Trans. 1 1990, 707–713.
- Umezaki, S.; Yokoshima, S.; Fukuyama, T. Total Synthesis of Lysergic Acid. Org. Lett. 2013, 15, 4230–4233.
- Crimmins, M.T.; King, B.W.; Tabet, E.A.; Chaudhary, K. Asymmetric Aldol Additions: Use of Titanium Tetrachloride and (−)-Sparteine for the Soft Enolization of N-Acyl Oxazolidinones, Oxazolidinethiones, and Thiazolidinethiones. J. Org. Chem. 2001, 66, 894–902.
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-Dihydroimidazol-2-Ylidene Ligands. Org. Lett. 1999, 1, 953–956.
- Kanno, R.; Yokoshima, S.; Kanai, M.; Fukuyama, T. Total Synthesis of (+)-Lysergic Acid. J. Antibiot. 2018, 71, 240–247.
- Evans, D.A.; Ennis, M.D.; Mathre, D.J. Asymmetric Alkylation Reactions of Chiral Imide Enolates. A Practical Approach to the Enantioselective Synthesis of Alpha.-Substituted Carboxylic Acid Derivatives. J. Am. Chem. Soc. 1982, 104, 1737–1739.
- Liu, Q.; Jia, Y. Total Synthesis of (+)-Lysergic Acid. Org. Lett. 2011, 13, 4810–4813.
- Liu, Q.; Zhang, Y.-A.; Xu, P.; Jia, Y. Total Synthesis of (+)-Lysergic Acid. J. Org. Chem. 2013, 78, 10885–10893.
- González-Gómez, J.C.; Medjahdi, M.; Foubelo, F.; Yus, M. Stereoselective α-Aminoallylation of Aldehydes with Chiral Tert-Butanesulfinamides and Allyl Bromides. J. Org. Chem. 2010, 75, 6308–6311.
- Oppolzer, W.; Grayson, J.I. Stereoselective Total Syntheses of (±)-Chanoclavine I and (±)-Isochanoclavine I by an Intramolecular Nitrone-Olefin/Cycloaddition. Preliminary Communication. Helv. Chim. Acta 1980, 63, 1706–1710.
- Plieninger, H.; Wagner, C.; Immel, H. Synthese von 4--Tryptophan- Sowie -Tryptamin- Und Einbau in Clavin-Alkaloide. Justus Liebigs Ann. Chem. 1971, 743, 95–111.
- Inuki, S.; Oishi, S.; Fujii, N.; Ohno, H. Total Synthesis of (±)-Lysergic Acid, Lysergol, and Isolysergol by Palladium-Catalyzed Domino Cyclization of Amino Allenes Bearing a Bromoindolyl Group. Org. Lett. 2008, 10, 5239–5242.
- Iwata, A.; Inuki, S.; Oishi, S.; Fujii, N.; Ohno, H. Formal Total Synthesis of (+)-Lysergic Acid via Zinc(II)-Mediated Regioselective Ring-Opening Reduction of 2-Alkynyl-3-Indolyloxirane. J. Org. Chem. 2011, 76, 5506–5512.
- Inuki, S.; Iwata, A.; Oishi, S.; Fujii, N.; Ohno, H. Enantioselective Total Synthesis of (+)-Lysergic Acid, (+)-Lysergol, and (+)-Isolysergol by Palladium-Catalyzed Domino Cyclization of Allenes Bearing Amino and Bromoindolyl Groups. J. Org. Chem. 2011, 76, 2072–2083.
- Kimura, M.; Futamata, M.; Mukai, R.; Tamaru, Y. Pd-Catalyzed C3-Selective Allylation of Indoles with Allyl Alcohols Promoted by Triethylborane. J. Am. Chem. Soc. 2005, 127, 4592–4593.
- Hofmeister, H.; Annen, K.; Laurent, H.; Wiechert, R. A Novel Entry to 17α-Bromo-and 17α-Iodoethynyl Steroids. Angew. Chem. Int. Ed. Engl. 1984, 23, 727–729.
- Myers, A.G.; Zheng, B. New and Stereospecific Synthesis of Allenes in a Single Step from Propargylic Alcohols. J. Am. Chem. Soc. 1996, 118, 4492–4493.
- Lauchli, R.; Shea, K.J. A Synthesis of the Welwistatin Core. Org. Lett. 2006, 8, 5287–5289.
- Uenishi, J.; Kawahama, R.; Shiga, Y.; Yonemitsu, O.; Tsuji, J. A General and Convenient Synthetic Method of Geometrically Pure (Z)-1-Bromo-1-Alkenes. Tetrahedron Lett. 1996, 37, 6759–6762.
- Movassaghi, M.; Ahmad, O.K. N -Isopropylidene- N’-2-Nitrobenzenesulfonyl Hydrazine, a Reagent for Reduction of Alcohols via the Corresponding Monoalkyl Diazenes. J. Org. Chem. 2007, 72, 1838–1841.
- Marino, J.P.; Osterhout, M.H.; Padwa, A. An Approach to Lysergic Acid Utilizing an Intramolecular Isomuenchnone Cycloaddition Pathway. J. Org. Chem. 1995, 60, 2704–2713.
- Padwa, A.; Austin, D.J. Ligand Effects on the Chemoselectivity of Transition Metal Catalyzed Reactions Ofα-Diazo Carbonyl Compounds. Angew. Chem. Int. Ed. Engl. 1994, 33, 1797–1815.
- Barton, D.H.R.; McCombie, S.W. A New Method for the Deoxygenation of Secondary Alcohols. J. Chem. Soc. Perkin Trans. 1 1975, 1574–1585.
- Parsons, P.J.; Penkett, C.S.; Shell, A.J. Tandem Reactions in Organic Synthesis: Novel Strategies for Natural Product Elaboratoin and the Development of New Synthetic Methodology. Chem. Rev. 1996, 96, 195–206.
- Cladingboel, D.E.; Parsons, P.J. Synthesis of Lysergic Acid Derivatives by Triple Radical Cyclisation. J. Chem. Soc. Chem. Commun. 1990, 1543–1544.
- Özlü, Y.; Cladingboel, D.E.; Parsons, P.J. Tandem Radical Cyclisations: Synthesis of Lysergic Acid Derivatives. Tetrahedron 1994, 50, 2183–2206.
- Floss, H.G. Biosynthesis of Ergot Alkaloids and Related Compounds. Tetrahedron 1976, 32, 873–912.
- Mukherjee, J.; Menge, M. Progress and Prospects of Ergot Alkaloid Research. In New Products and New Areas of Bioprocess Engineering. Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2000; Volume 68, pp. 1–20.
- Tsai, H.F.; Wang, H.; Gebler, J.C.; Poulter, C.D.; Schardl, C.L. The Claviceps Purpurea Gene Encoding Dimethylallyltryptophan Synthase, the Committed Step for Ergot Alkaloid Biosynthesis. Biochem. Biophys. Res. Commun. 1995, 216, 119–125.
- Lee, S.-L.; Floss, H.G.; Heinstein, P. Purification and Properties of Dimethylallylpyrophosphate: Tryptophan Dimethylallyl Transferase, the First Enzyme of Ergot Alkaloid Biosynthesis in Claviceps Sp. SD 58. Arch. Biochem. Biophys. 1976, 177, 84–94.
- Metzger, U.; Schall, C.; Zocher, G.; Unsöld, I.; Stec, E.; Li, S.-M.; Heide, L.; Stehle, T. The Structure of Dimethylallyl Tryptophan Synthase Reveals a Common Architecture of Aromatic Prenyltransferases in Fungi and Bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 14309–14314.
- Rigbers, O.; Li, S.-M. Ergot Alkaloid Biosynthesis in Aspergillus Fumigatus. J. Biol. Chem. 2008, 283, 26859–26868.
- Tudzynski, P.; Correia, T.; Keller, U. Biotechnology and Genetics of Ergot Alkaloids. Appl. Microbiol. Biotechnol. 2001, 57, 593–605.
- Lorenz, N.; OlŠovská, J.; šulc, M.; Tudzynski, P. Alkaloid Cluster Gene CcsA of the Ergot Fungus Claviceps Purpurea Encodes Chanoclavine I Synthase, a Flavin Adenine Dinucleotide-Containing Oxidoreductase Mediating the Transformation of N-Methyl-Dimethylallyltryptophan to Chanoclavine I. Appl. Environ. Microbiol. 2010, 76, 1822–1830.
- Goetz, K.E.; Coyle, C.M.; Cheng, J.Z.; O’Connor, S.E.; Panaccione, D.G. Ergot Cluster-Encoded Catalase Is Required for Synthesis of Chanoclavine-I in Aspergillus Fumigatus. Curr. Genet. 2011, 57, 201–211.
- Ryan, K.; Moore, C.; Panaccione, D. Partial Reconstruction of the Ergot Alkaloid Pathway by Heterologous Gene Expression in Aspergillus Nidulans. Toxins 2013, 5, 445–455.
- Wallwey, C.; Matuschek, M.; Li, S.-M. Ergot Alkaloid Biosynthesis in Aspergillus Fumigatus: Conversion of Chanoclavine-I to Chanoclavine-I Aldehyde Catalyzed by a Short-Chain Alcohol Dehydrogenase FgaDH. Arch. Microbiol. 2010, 192, 127–134.
- Cheng, J.Z.; Coyle, C.M.; Panaccione, D.G.; O’Connor, S.E. Controlling a Structural Branch Point in Ergot Alkaloid Biosynthesis. J. Am. Chem. Soc. 2010, 132, 12835–12837.
- Matuschek, M.; Wallwey, C.; Xie, X.; Li, S.-M. New Insights into Ergot Alkaloid Biosynthesis in Claviceps Purpurea: An Agroclavine Synthase EasG Catalyses, via a Non-Enzymatic Adduct with Reduced Glutathione, the Conversion of Chanoclavine-I Aldehyde to Agroclavine. Org. Biomol. Chem. 2011, 9, 4328–4335.
- Kim, S.-U.; Cho, Y.-J.; Floss, H.; Anderson, J. Conversion of Elymoclavine to Paspalic Acid by a Particulate Fraction from an Ergotamine-Producing Strain of Claviceps sp. Planta Med. 1983, 48, 145–148.
- Maier, W.; Schumann, B.; Gröger, D. Microsomal Oxygenases Involved in Ergoline Alkaloid Biosynthesis of VariousClaviceps Strains. J. Basic Microbiol. 1988, 28, 83–93.
- Britton, K.N.; Steen, C.R.; Davis, K.A.; Sampson, J.K.; Panaccione, D.G. Contribution of a Novel Gene to Lysergic Acid Amide Synthesis in Metarhizium Brunneum. BMC Res. Notes 2022, 15, 183.
- Chen, J.-J.; Han, M.-Y.; Gong, T.; Yang, J.-L.; Zhu, P. Recent Progress in Ergot Alkaloid Research. RSC Adv. 2017, 7, 27384–27396.
- Robinson, S.L.; Panaccione, D.G. Heterologous Expression of Lysergic Acid and Novel Ergot Alkaloids in Aspergillus Fumigatus. Appl. Environ. Microbiol. 2014, 80, 6465–6472.
- Wong, G.; Lim, L.R.; Tan, Y.Q.; Go, M.K.; Bell, D.J.; Freemont, P.S.; Yew, W.S. Reconstituting the Complete Biosynthesis of D-Lysergic Acid in Yeast. Nat. Commun. 2022, 13, 712.
This entry is offline, you can click here to edit this entry!