Mitochondria are double membrane organelles that harbor their own DNA (mtDNA). They are often referred to as the powerhouse of the cell as they produce adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS). The OXPHOS pathway generates ATP by several oxidation-reduction reactions involving electron transfer from NADH and FADH
2 to oxygen across transmembrane protein complexes in the inner mitochondrial membrane. In these reactions, NADH and FADH
2 are oxidized to NAD
+ and FAD, respectively [
1]. Apart from ATP production, mitochondria also play essential roles in various other cellular functions such as intracellular calcium (Ca
2+) homeostasis and regulation of apoptosis [
2]. Neurons critically depend on mitochondria due to their high energy demand and need for tight Ca
2+ regulation to maintain neuronal activity [
3]. Consequently, mitochondrial dysfunction is directly linked to neurodegenerative diseases and aging [
4,
5]. Neurons are highly polarized cells with a complex structure and different subcellular compartments including a cell body, dendrites, axons and synapses. The different parts of the neuron do not only have different functions but also different energy demands. As a consequence, a finely tuned mitochondrial network constantly adapting to the local requirement of the neuronal compartment is necessary. This is achieved by mitochondrial transport along axons and dendrites, fusion and fission and degradation of damaged organelles via mitophagy, as well as mitochondrial biogenesis [
6,
7]. As mitochondria cannot be made de novo, the biogenesis of mitochondrial proteins requires already existing organelles, which duplicate and express their mtDNA, as well as import of over 1000 proteins encoded in the nucleus [
8].
2. Regulation of Mitochondrial Biogenesis by PGC-1α
The co-transcriptional regulation factor PGC-1α (peroxisome-proliferator-activated γ co-activator-1α) is known as the master regulator of mitochondrial biogenesis. It activates different transcription factors including NRF-1 and NRF-2 (nuclear respiration factors 1 and 2), which promote the expression of several nuclear-encoded mitochondrial genes. Furthermore, they drive the expression of the nuclear-encoded TFAM (mitochondrial transcription factor A), which is required for transcription and replication of mtDNA [
9,
10].
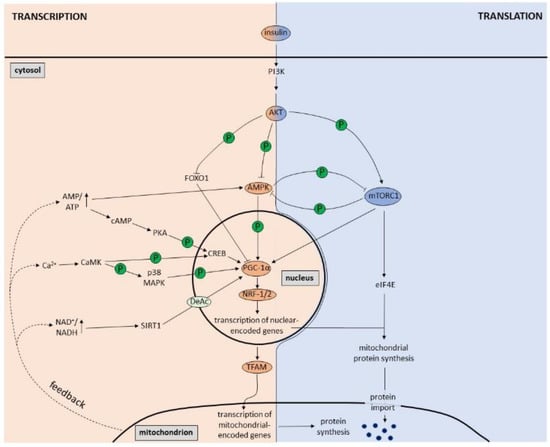
Several intracellular signaling molecules generated or regulated by mitochondria (
Figure 1) are involved in promoting mitochondrial biogenesis via the PGC-1α-NRF-1/2-TFAM pathway, including the AMP/ATP ratio (via AMPK), the NAD
+/NADH ratio (via SIRT1) and Ca
2+ levels (via CaMK) [
11]. AMPK (adenosine monophosphate-activated protein kinase) is a serine/threonine protein kinase that is composed of one catalytic α subunit and two regulatory β and γ subunits [
12]. An increased AMP/ATP ratio induces activation of AMPK, which directly phosphorylates and thereby activates PGC-1α [
13]. PGC-1α in turn controls expression of several mitochondrial genes as well as its own [
13,
14]. In addition to activating AMPK, AMP can also be converted to cyclic AMP (cAMP) by adenyl cyclase and subsequently stimulate PKA activity. PKA in turn phosphorylates the cAMP response element binding (CREB) protein in the nucleus [
15]. CREB is a transcription factor that binds to the promoter of PGC-1α [
16], thereby promoting mitochondrial biogenesis. Apart from AMPK, SIRT1 (Sirtuin 1) is another energy sensor that plays an important role in mitochondrial biogenesis. SIRT1 is a deacetylase that is activated in response to an increased NAD
+/NADH ratio caused by energy stress. Once activated, SIRT1 deacetylates PGC-1α resulting in its activation [
17]. Interestingly, the signaling pathways of the two major energy sensors, AMPK and SIRT1, seem to be interconnected as AMPK acts upstream of SIRT1 by increasing intracellular NAD
+ levels leading to SIRT1 activation, deacetylation of PGC-1α and mitochondrial biogenesis [
18,
19,
20,
21]. Finally, Ca
2+ also plays an important role in regulating mitochondrial biogenesis via the PGC-1α-NRF-1/2-TFAM pathway [
22]. Mechanistically, Ca
2+ promotes CaMK (calcium/calmodulin-dependent protein kinase) activity, which phosphorylates p38 MAPK (p38 mitogen-activated protein kinase), resulting in activation of PGC-1α [
22,
23,
24]. Interestingly, CaMK can also stimulate PGC-1α via CREB activation [
14]. Consequently, CREB might be involved in both AMP- and Ca
2+-dependent mitochondrial biogenesis.
Figure 1. Regulation of mitochondrial biogenesis at the transcriptional and translational level. At the transcriptional level, the PGC-1α-NRF-1/2-TFAM pathway is the master regulator of mitochondrial biogenesis. Increased AMP/ATP ratio, NAD+/NADH ratio and Ca2+ levels, which are part of mitochondrial feedback mechanisms within the cell, result in activation of AMPK, PKA, SIRT1 and CaMK that in turn lead to PGC-1α stimulation. Once activated, PGC-1α promotes transcription of nuclear-encoded mitochondrial genes via NRF-1/2 and transcription of mitochondrial-encoded genes via expression of TFAM. At the translational level, the insulin-induced PI3K/AKT/mTORC1 pathway plays a major role in mitochondrial biogenesis. Via activation of eIF4E, mTORC1 increases the translation of nuclear-encoded mitochondrial proteins, which are imported into mitochondria. AKT, however, also has an effect on transcription via inhibition of FOXO1 and consequent activation of PGC-1α. Furthermore, AMPK is also a substrate of AKT, which is inhibited by AKT-induced phosphorylation. Finally, AMPK and mTORC1 can inhibit each other by direct phosphorylation.
Since the majority of the studies about mitochondrial biogenesis have been performed in non-neuronal cells, very little is known about its regulation in neurons. However, as in other cells, PGC-1α is the master regulator of mitochondrial biogenesis in neurons [
11]. Overexpression of PGC-1α has been shown to result in an increased number of mitochondria and improved mitochondrial function in primary hippocampal neurons [
25]. Knockdown of PGC-1α, on the other hand, leads to a reduction in dendritic mitochondria and inhibition of synaptogenesis in hippocampal neurons [
26]. Furthermore, PGC-1α has been shown to control axonal mitochondrial density in a SIRT1-dependent manner [
27]. It remains to be determined how much local activation of AMPK, SIRT1 or CaMK can elicit global changes in mitochondrial transcription as discussed below.
3. Regulation of Mitochondrial Biogenesis by Signaling Pathways
Insulin, the main hormone involved in fuel metabolism, is an important regulator of mitochondrial biogenesis (
Figure 1). Upon binding to the insulin receptor, insulin elicits the phosphoinositide 3-kinase (PI3K)-dependent activation of AKT [
28]. AKT in turn phosphorylates and thereby inhibits the transcription factor FOXO1 (Forkhead Box Protein O1) [
29,
30,
31]. AKT-induced FOXO1 inhibition increases expression and activity of PGC-1α thereby stimulating transcription of mitochondrial genes [
32,
33,
34,
35,
36]. Despite some conflicting studies [
37,
38], the overall agreement is that insulin signaling promotes mitochondrial biogenesis [
39,
40]. In line with this, several studies have shown that insulin treatment stimulates mitochondrial protein synthesis and function [
41,
42]. AKT also activates mTORC1 (mammalian target of rapamycin complex 1), which not only regulates mitochondrial oxidative function [
43] via stimulation of PGC-1α [
44] but also increases the translation of nuclear-encoded mitochondrial proteins via activation of eIF4E (eukaryotic translation initiation factor 4E) [
45]. Interestingly, insulin signaling is also clearly linked to various aspects of neuronal mitochondrial function including respiration, ATP production and Ca
2+ buffering as well as protein homeostasis and biogenesis [
46]. In cortical neurons, insulin treatment has been shown to enhance mitochondrial respiration [
47]. This in vitro finding could be confirmed in mice, where intranasal application of insulin increases brain mitochondrial respiration accompanied by enhanced mtDNA levels as well as increased mitochondrial protein levels and PGC-1α expression in the hippocampus [
48]. This supports the model that insulin signaling promotes mitochondrial biogenesis also in neurons.
To add to the complexity, the insulin/AKT pathway has also been shown to inhibit AMPK through phosphorylation via AKT [
49,
50,
51,
52,
53,
54,
55]; and mTORC1 directly inhibits AMPK through an inhibitory phosphorylation [
56]. AMPK, conversely, can also downregulate mTORC1 signaling via phosphorylation [
57,
58]. This reciprocal inhibition may result in a differential regulation of mitochondrial biogenesis at the transcriptional versus the translational level. Hence inhibition of mTORC1 signaling by AMPK would reduce mitochondrial protein biogenesis despite an increase in transcription and vice versa. This may explain how mitochondrial synthesis and activity can be decreased (via reduced mTORC1 signaling) despite increased expression of PGC-1α (via increased AMPK signaling). This is in line with several studies demonstrating that the correlation between mammalian expression levels of mRNA and protein is fairly low [
59,
60,
61] and that shifts in metabolism, as for example seen during neuronal differentiation, are executed at the translational level [
62].
Of note, the signals stimulating PGC-1α expression, including the AMP/ATP ratio, the NAD+/NADH ratio and Ca2+, are part of mitochondrial feedback mechanisms within the cell (Figure 1). Cell-intrinsic signals may demand a more long-term and therefore slower adaptation, which is obtained by transcriptional upregulation of mitochondrial genes, in contrast to the quick and transient regulation of translation by mTORC1 triggered by extrinsic signals such as insulin and other growth factors.
Finally, also other steps beyond transcription and translation could be controlled by cellular signaling pathways. This includes the localization of mRNAs encoding mitochondrial proteins (as discussed below) and the regulated removal of mitochondria by selective autophagy processes (mitophagy). In mammals, several mitophagy pathways and proteins have been described including PINK1 (PTEN-induced kinase 1), Parkin, BNIP3L/NIX and FUNDC1 [
63]. The PINK1/Parkin-dependent pathway is the best characterized pathway for degradation of damaged mitochondria. Briefly, in healthy mitochondria, PINK1 needs to be constantly synthesized, imported and degraded [
64]. On damaged mitochondria, however, PINK1 is stabilized and recruits Parkin to the mitochondria, which in turn leads to initiation of mitophagy, recruitment of autophagy receptors and eventually lysosomal degradation [
65,
66]. Similar to mitochondrial biogenesis, mitophagy is also regulated by cellular signaling pathways including AMPK signaling (see [
67] for more detail). Interestingly, in addition to their well-known role in mitophagy, PINK1 and Parkin are also involved in regulating mitochondrial biogenesis (as discussed below). Additionally, the import of mitochondrial precursor proteins through the translocases of the outer (TOM) and inner membrane was found to be regulated by several kinases in yeast [
8,
68,
69,
70,
71] and recently also confirmed to be regulated in mammalian cells [
72]. It will be interesting to reveal further mechanistic connections between these processes and the transcriptional as well as translational pathways described here.