Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Antipsychotic drugs have numerous disabling side effects, and many are lipophilic, making them hard to formulate at high strength. Incorporating them into nanometric emulsions can increase their solubility, protect them from degradation, and increase their brain delivery, being a promising strategy to overcome the current treatment gap.
- antipsychotic
- brain delivery
- microemulsion
- nanoemulsion
1. Schizophrenia: Pathophysiology, Current Therapy and Treatment Gap
Schizophrenia is a high incidence psychiatric disorder, having been recently estimated to affect 21 million people worldwide [1]. Its onset usually occurs in early adulthood and has a high impact on a patients’ quality of life, since it is characterized by a set of symptoms that not only affect the individual’s day-to-day functioning, but also their surroundings [1,2]. These symptoms include: hallucinations, delusions, disorganized thought or speech, and unusual behavior as positive symptoms; reduced motivation, lack of enjoyment, social withdrawal and flattened emotions as negative symptoms; and memory and attention deficits as cognitive symptoms [2,3]. Although the exact pathophysiology of schizophrenia remains unclear, disruptions in several neurotransmitter systems (serotonergic, cholinergic, glutamatergic, GABAergic, dopaminergic) have been associated with the disease [1,2,3]. However, the most affected neurotransmitter system in schizophrenic patients seems to be the dopaminergic system, with the existence of a dysfunction in dopamine production, which either increases (associated with positive symptoms) or decreases (associated with negative and cognitive symptoms) (summary of the pathophysiology of schizophrenia in Figure 1).
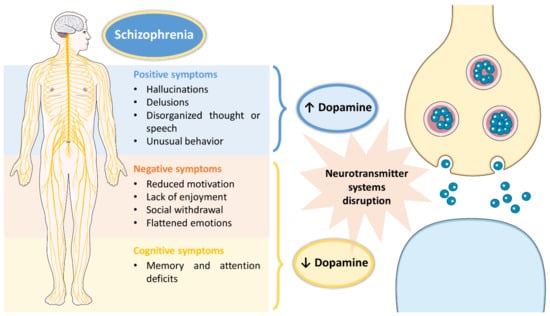
Figure 1. Pathophysiology of schizophrenia, including associated symptoms and pathological mechanisms at the cellular level.
Therefore, the chosen pharmacological treatment in a clinical context are usually antipsychotic drugs that mostly target dopamine receptors. Dozens of different drugs are available in the market and, hence, the treatment has to be tailored to the individual [1,2,4]. Antipsychotics can also be used to treat other schizoaffective or delusional disorders (alone or in combination with other drugs), such as acute mania, major depressive disorder with psychotic features, delusional disorder, severe agitation, Tourette syndrome, borderline personality disorder, dementia and delirium [5]. Nevertheless, in general, antipsychotic drugs lead to a large number of disabling side effects (metabolic, cardiovascular, neurologic), and many times result in relapse and treatment resistance. These limitations many times lead to treatment discontinuation [1,3]. Consequently, safer and more effective treatment options are urgently required. Yet, since many of these molecules have limiting aqueous solubility issues, due to being highly lipophilic, they are very hard to formulate, in technological terms.
2. Potential of Nanosystems for Brain Drug Delivery
Nanosystems, whose colloidal structure has a mean diameter of less than 500 nm, are drug delivery systems that have been showing a high number of advantages for biomedical applications. Aside from increasing drugs’ solubilization and protecting them from degradation (both metabolic and chemical), these structures increase drug transport through biological membranes and, ultimately, promote improved brain bioavailability. Since the very low permeability of the blood-brain barrier blocks the entrance of the grand majority of molecules (especially hydrophilic and/or high molecular weight compounds), this performance is essential for diseases with a brain etiology, in which brain targeting is required, being thereby capable of decreasing the systemic bioavailability, prompting to increased safety due to less systemic side effects, while increasing the therapeutic efficacy [6,7,8,9,10]. There are several types of nanosystems, with the main categories being: polymeric nanosystems (polymeric nanoparticles, polymeric micelles), lipid nanoparticles (solid lipid nanoparticles, nanostructured lipid carriers), liposomes and related nanosystems (transfersomes, cubosomes, ethosomes), and nanometric emulsions [11]. Nevertheless, most of these systems have been reported to have short/low physical stability, low encapsulation efficiency, and non-biocompatible components, while requiring complex preparation methods, that are time- and resource-consuming, and that frequently use toxic organic solvents [12,13,14]. Yet, nanometric emulsions, which consist of colloidal liquid-in-liquid dispersions, can form spontaneously, evidencing the great advantage of having straightforward methods of preparation, that are not quite common. They can also be prepared by high-energy methods (such as by using high-pressure homogenizers, ultrasonication, or extrusion through a small pore synthetic membrane), but spontaneous emulsification is possible when its components are in the right proportions. Moreover, their preparation does not require the use of toxic organic solvents. They are made of a water phase, oils, surfactants, and cosurfactants or cosolvents, and can be classified according to the nature of their internal and external phases, being divided into oil-in-water (O/W) or water-in-oil nanometric emulsions. Moreover, they can also be classified according to their droplet size, being divided into microemulsions (10–100 nm) or nanoemulsions (20–200 nm) (although the accepted size range can differ between authors). Aside from the possibility of having simple and fast preparation methods, nanometric emulsions also have all of the aforementioned biopharmaceutical advantages of nanosystems, and their lipophilic nature makes them ideal for solubilizing lipophilic drugs. They are also biocompatible, and the right formulas can be quite stable. Additionally, the surfactants and cosolvents that are part of these formulations composition have the ability to enhance drug permeation across biological barriers [11,15,16] (summary of these characteristics in Figure 2).
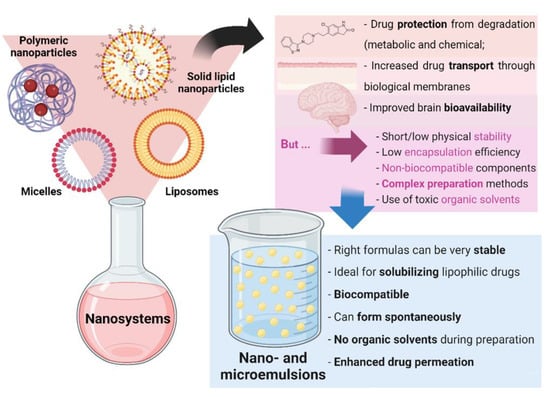
Figure 2. General nanosystem types and their characteristics, as well as the advantages of nano and microemulsions.
This review was projected with the purpose of collecting and analyzing detailed relevant information regarding nano- and micro-emulsions development for the delivery of antipsychotic drugs. This comprehensive information includes composition, droplet size, polydispersity index (PDI), zeta potential, viscosity, osmolarity/osmolality, pH, in vitro drug release, ex vivo drug permeation, in vivo pharmacokinetics, in vivo pharmacodynamics and/or safety studies, depending on the available original data. The final goal was to address what has been performed so far in this specific scientific pharmaceutical and medical field, while attempting to conclude on which nanometric emulsion composition and/or incorporated drug(s) could be most adequate for the therapeutic aim.
3. First-Generation Antipsychotics
First-generation (or typical) antipsychotics are effective in attenuating schizophrenia’s positive symptoms, mainly associated with acute episodes, and preventing psychotic relapse [4].
Chlorpromazine was the first antipsychotic drug to ever be approved for clinical application, with its use spreading worldwide between 1952 and 1955 [38]. It is mostly a dopamine and serotonin receptor antagonist, but it also has affinity for other receptors, which leads to several severe systemic side effects, including agitation, convulsions, difficulty breathing and swallowing, extreme sleepiness, fever, irregular heart rate, and low blood pressure [17,39]. Its lipophilic nature leads to it evidencing both poor aqueous solubility (predictably 0.00417 mg/mL) and poor permeability, which in turn results in variable and low oral absorption. Moreover, chlorpromazine holds a high protein-binding and extensive first-pass metabolism. These characteristics are responsible of a low oral bioavailability [17,39]. To tackle these issues, Baloch et al. formulated chlorpromazine into a self-nanoemulsifying drug delivery system (SNEDDS). SNEDDS are mixtures of oils, surfactants and cosurfactants or cosolvents that spontaneously form a nanoemulsion when in contact with (diluted in) the gastrointestinal tract’s fluids [40]. All 3 formulas comprised Tween® 85 (polysorbate 85-polyoxyethylene 20 sorbitan trioleate) as the hydrophilic surfactant and ethanol as cosolvent, and 2 of the formulas also had glycerol. The selected oil was different among the developed formulations, being either Triacetin (short-chain triglyceride), Captex® 355 (medium-chain triglyceride, glyceryl tricaprylate/tricaprate) or olive oil + linseed oil (long-chain triglycerides, 1:2 w/w). Detailed quantities of each used ingredient are shown in Table 1. Drug strength was kept at 2% (w/w) for all formulations, which is 4796 times higher than chlorpromazine’s predicted aqueous solubility. SNEDDS’ droplet characteristics (after dilution) were 159–186 nm for droplet size, 0.27–0.33 for PDI, and −14.1 to −21.4 mV for zeta potential, with a pH between 7 and 7.5. Moreover, all SNEDDS were stable under accelerated conditions or storage for 3 months. Ex vivo permeation studies (rat intestine) showed that all SNEDDS had increased drug permeation when compared to a drug suspension, which was justified by the tight junctions’ opening effect of triglycerides that facilitates drug transport across the intestinal mucosa. Moreover the long-chain triglyceride formula showed the best performance, which suggests that the presence of oleic acid had a permeation enhancing effect. The in vivo pharmacokinetic studies (with rats; 2 mg drug/kg body weight) showed that the oral administration of all SNEDDS led to an at least 2-fold higher systemic bioavailability than the free drug suspension. The presence of a longer chain triglyceride seemed to improve the formulation’s performance once more, being responsible for the highest plasma area under the “drug concentration vs. time” curve (AUC) and maximum drug concentration (Cmax). Nevertheless, there was no intravenous control group, which would have provided important data regarding the drug’s systemic bioavailability. Moreover, the drug was never quantified in the animals’ brain, nor was a pharmacodynamic study performed, and hence the potential of the developed formulations to allow the drug to reach the brain and/or have a therapeutic effect was left undetermined.
Table 1. Detailed composition of chlorpromazine and haloperidol nanometric emulsions. Excipient and drug quantities and units are shown as reported in the respective articles (w/w% for excipients, and w/w% or mg/mL for drugs). For the excipients the brand name was used (when available).
| Composition | Oral Chlorpromazine SNEDDS [17] (% w/w) | Intranasal Haloperidol O/W Nanoemulsions [18] | ||||
|---|---|---|---|---|---|---|
| With Short-Chain Triglyceride | With Medium-Chain Triglyceride | With Long-Chain Triglyceride | Non-Mucoadhesive | Mucoadhesive | ||
| Oil | Triacetin | 40% | - | - | - | - |
| Captex® 355 1 | - | 35% | - | - | - | |
| Olive oil + linseed oil | - | - | 55% | - | - | |
| Capmul® MCM 2 | - | - | - | NR | NR | |
| Hydrophobic surfactant | Span® 20 3 | - | - | - | NR | NR |
| Hydrophilic surfactant | Tween® 85 4 | 48% | 50% | 40% | - | - |
| Tween® 80 5 | - | - | - | NR | NR | |
| Cosolvent | Ethanol | 5% | 5% | 3% | - | - |
| Glycerol | 5% | 8% | - | - | - | |
| Transcutol® P 6 | - | - | - | NR | NR | |
| Mucoadhesive and/or viscosifying agents | Pemulen™ TR-2 7 | - | - | - | - | 0.3% w/w |
| Aqueous phase | Water | - | - | - | NR | NR |
| Drug | Chlorpromazine | 2% | 2% | 2% | - | - |
| Haloperidol | - | - | - | 8.5 mg/mL | 8.5 mg/mL | |
Haloperidol is a strong dopamine D2 receptor antagonist that was approved for clinical use in the 1960s [18,41,42]. It is currently available in oral and injectable drug preparations, but it has limited uptake across the blood brain-barrier, and a vast distribution to non-targeted sites, which leads to quite severe side effects, such as movement disorders (dystonia, tardive dyskinesia, akathisia and drug-induced Parkinsonism), sedation, weight gain, prolactin changes and cardiac events. Moreover, haloperidol undergoes high liver metabolization, resulting in low oral bioavailability, and low aqueous solubility (predictably 0.00446 mg/mL) [18,42]. In order to overcome such limitations, El-Setouhy et al. developed O/W nanoemulsions for intranasal delivery, containing Capmul® MCM (medium chain mono- and di-glycerides), Tween® 80 (polysorbate 80-polyoxyethylene 20 sorbitan monooleate), Span 20 (sorbitan monolaurate) and Transcutol® P (diethylene glycol monoethyl ether), prepared by spontaneous emulsification. Additionally, the mucoadhesive nanoemulsion also contained Pemulen™ TR-2 (acrylates/C10–30 alkyl acrylate crosspolymer) (specific quantity in Table 2). Obtained drug strength was 8.5 mg/mL, which is more than 1900 times higher than haloperidol’s aqueous solubility. For the selected non-mucoadhesive formula, droplet size was 209.5 nm, but PDI was 0.402, which is a reasonably high value, hence reflecting the non-homogeneous nature of the formulation, while zeta potential was found to be −23.85 mV. This same formula was stable after a 12-month storage period (both under refrigeration and at room temperature). However, the mucoadhesive nanoemulsion was not characterized, which raises the question of whether it was stable, homogeneous, and even if it was in fact a nanoemulsion. The in vivo pharmacokinetic study (mice; 2 mg/kg), which was only performed for the mucoadhesive nanoemulsion, (which was not the one that was previously characterized), showed that its intranasal administration led to a more effective and faster brain drug delivery (higher brain Cmax, with similar AUC, and lower “time to reach maximum drug concentration” (Tmax)), suggesting the superiority of this route of administration for this drug delivery purpose. Additionally, intranasal delivery led to brain/blood ratios that were higher than 1 at all time-points (which did not happen with the intravenous group), suggesting higher efficacy and safety (lower desirable systemic distribution). The fact that the drug was located within lipophilic nanodroplets might have facilitated the direct nasal transmucosal transport to the brain, in which the presence of the mucoadhesive Pemulen™ TR2 helped overcome nasal mucociliary clearance. Nevertheless, a mucoadhesive nanoemulsion is hardly an ideal intravenous control, especially if it is too viscous (an issue that was not addressed in this study), and the intravenous administration of a drug solution would have been a more adequate control, also providing a comparison with a different and simpler formulation. Additionally, the administration of the same formulation through two different routes did not allow us to understand whether the formulation itself was more effective or safer than others (drug solutions or suspensions), but only that its intranasal administration was more effective in making the drug reach the brain than the intravenous. Still, the in vivo pharmacodynamic study (locomotor activity) further confirmed the effectiveness of the developed intranasal mucoadhesive nanoemulsion, which was very effective in causing motor suppression. Safety evaluation (rabbits; 0.5 mg/kg) suggested that the developed formulation was also safe, with the animals’ nasal mucosa showing no histopathological alterations after chronic administration.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics14102174
This entry is offline, you can click here to edit this entry!