Monkeypox virus (MPXV) is an adenovirus with a double-stranded DNA genome, belonging to the family Poxviridae, subfamily Chordopoxvirinae, and the genus Orthopoxvirus. MPXV was first reported in 1958 after two pox-like disease outbreaks occurred in monkeys. The original source of MPXV is unknown. Rodents likely harbor the virus, leading to spillover events. The case of human infection by MPXV was first reported in humans in in the Democratic Republic of the Congo in 1970.
- monkeypox
- real-time PCR
- immunoassay
- diagnosis
1. Indirect Detection
1.1. Monkeypox Diagnosis Based on Virus Culture
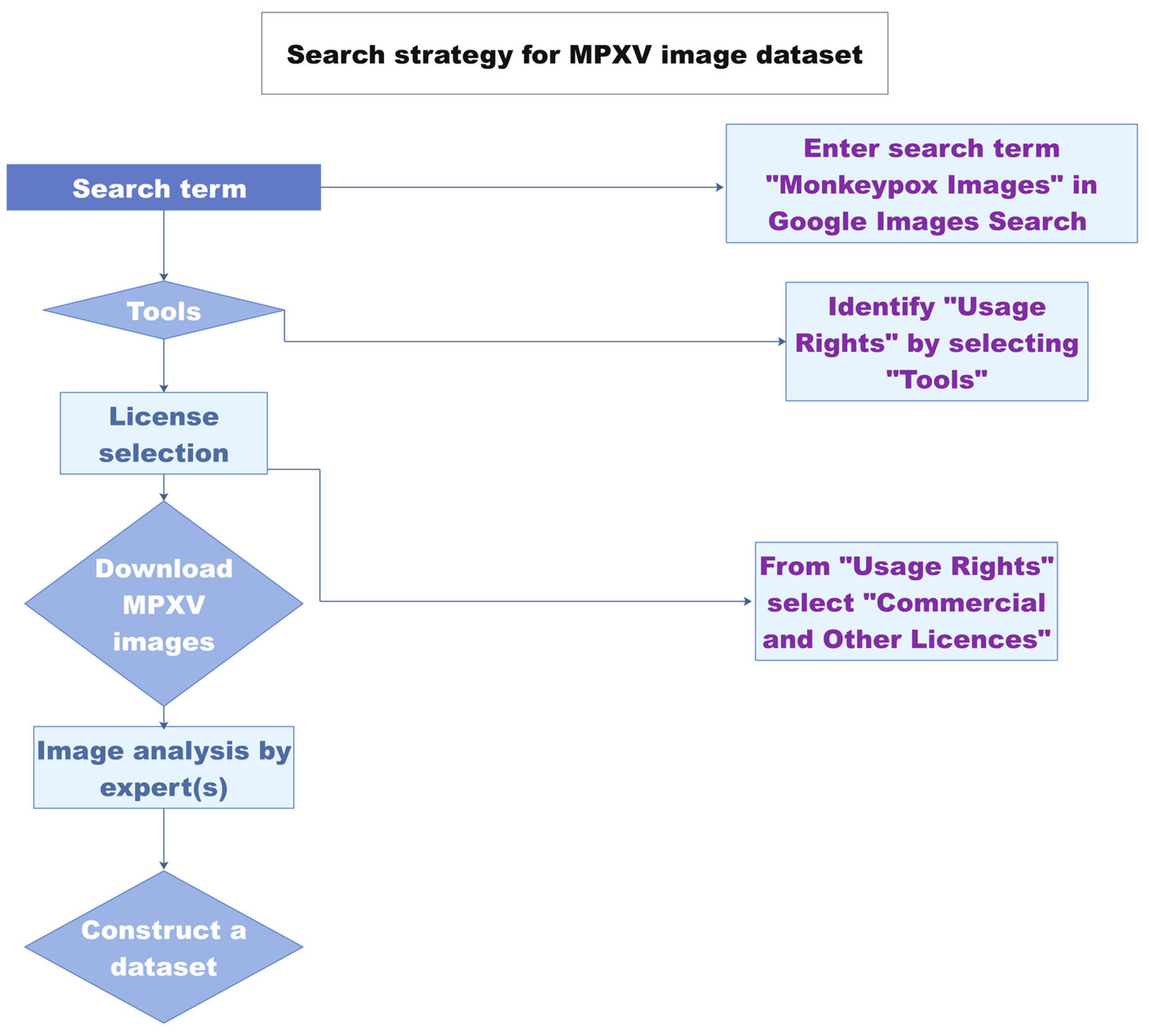
1.2. Diagnosis Based on Image Analysis
2. Direct Detection
2.1. Monkeypox Immunodiagnostics
2.2. Whole-Particle Detection
2.3. Detection by Genome Sequencing
2.4. Monkeypox Virus Detection Based on PCR
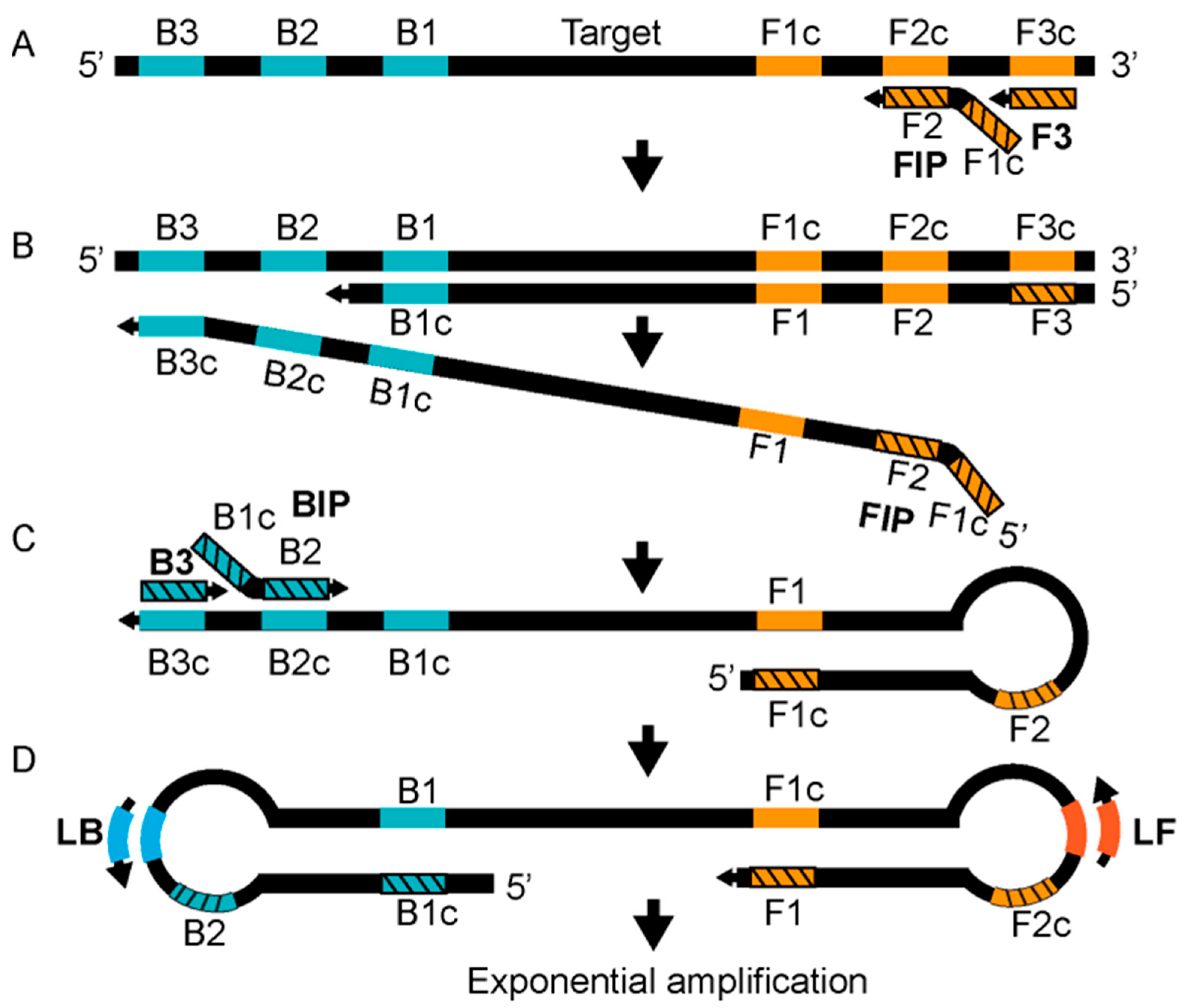
2.5. Detection Based on Isothermal Amplification
|
Sr. No |
Assay Name |
Target Gene |
Primers’ Sequences |
Probes’ Sequences |
Detection Limit |
Real-Sample Analysis |
References |
|---|---|---|---|---|---|---|---|
|
1 |
HA-PCR |
HA gene |
Forward: 5′-CTGATAATGTAGAAG AC -3′ Reverse: 5′-TTGTATTTACGTGGGTG-3′ |
NA |
Not reported |
Yes |
[25] |
|
2 |
ATI-PCR |
ATI-gene |
Forward: 5′-AATACAAGGAGGATCT-3′ Reverse: 5′-CTTAACTTTTTCTTTTTCTTTCTC-3′ |
NA |
Not reported |
Yes |
[26] |
|
3 |
MPXV PCR assay |
ATI-gene |
Forward: 5′-GAGAGAATCTCTTGATAT-3′ Reverse: 5′-ATTCTAGATTGTAATC-3′ |
NA |
Not reported |
Yes |
[28] |
|
4 |
Real-time PCR |
B6R |
Forward: 5′-ATTGGTCATTATTTTTGTCACAGGAACA-3′ Reverse: 5′-AATGGCGTTGACAATTATGGGTG-3′ |
5′-MGB/DarkQuencher-AGAGATTAGAAATA-3′-FAM |
∼10 viral copies (2 fg) |
Yes |
[29] |
|
5 |
Real-time PCR |
G2R |
Forward: 5′-CACACCGTCTCTTCCACAGA -3′ Reverse: 5′-GATACAGGTTAATTTCCACATCG -3′ |
5′-FAM AACCCGTCGTAACCAGCAATACATTT-3′-BHQ1 |
∼8.2 genome copies (1.7 fg) |
Yes |
[47] |
|
6 |
Real-time PCR |
G2R |
Forward: 5′-TGTCTACCTGGATACAGAAAGCAA-3′ Reverse: 5′-GGCATCTCCGTTTAATACATTGAT -3′ |
5′-FAM-CCCATATATGCTAAATGTACCGGTACCGGA-3′-BHQ1 |
∼40.4 copies (9.46 fg) |
Yes |
[47] |
|
7 |
Real-time PCR |
F3L |
Forward: 5′-CTCATTGATTTTTCG CGGGAT A-3′ Reverse: 5′-GACGATACTCCTCCT CGTTGGT-3′ |
5′-6FAM-CATCAGAATCTGTAGGCCGT-MGBNFQ-3′ |
11–55 fg (50–250 copies) |
Yes |
[48] |
|
8 |
Real-time PCR |
N3R |
Forward: 5′-AACAACCGT CCTACA ATTAAA CAACA-3′ Reverse: 5′-CGCTATCGAACCATT TTTGTAGTCT-3′ |
5′-6FAM-TAT AAC GGC GAA GAA TAT ACT-MGBNFQ-3′ |
11–55 fg (50–250 copies) |
Rodents |
[48] |
|
9 |
Real-time PCR |
B7R |
Forward: 5′-ACGTGTTAAACAATGGGTGATG-3′ Reverse: 5′-AACATTTCCATGAATCGTAGTCC-3′ |
5′-TAMRA-TGAATGAATGCGATACTGTATGTGTGGG-3′-BHQ2 |
50 copies per reaction |
Yes |
[49] |
|
10 |
C-LAMP |
D14L |
FIP-C: 5′-TGGGAGCATTGTAACTTATAGTTGCCCTCCTGAACACATGACA-3′ F3-C: 5′-TGGGTGGATTGGACCATT-3′ BIP-C: 5′-ATCCTCGTATCCGTTATGTCTTCCCACCTATTTGCGAATCTGTT-3′ B3-C: 5′-ATGGTATGGAATCCTGAGG-3′ LOOP-F-C: 5′-GATATTCGTTGATTGGTAACTCTGG-3′ LOOP-C-C: 5′-GTTGGATATAGATGGAGGTGATTGG-3′ |
N/A |
102.4 copies per reaction |
Yes |
[42] |
|
11 |
C-LAMP |
ATI |
FIP-W: 5′-CCGTTACCGTTTTTACAATCGTTAATCAATGCTGATATGGAAAAGAGA-3′ F3-W: 5′-TACAGTTGAACGACTGCG-3′ BIP-W: 5′-ATAGGCTAAAGACTAGAATCAGGGATTCTGATTCATCCTTTGAGAAG-3′ B3-W: 5′-AGTTCAGTTTTATATGCCGAAT-3′ LOOP-F-W: 5′-GATGTCTATCAAGATCCATGATTCT-3′ LOOP-C-W: 5′-TCTTGAACGATCGCTAGAGA-3′ |
N/A |
103 copies per reaction |
Yes |
[42] |
|
12 |
RPA |
G2R |
Forward: 5′-AATAAACGGAAGAGATATAGCACCACATGCAC-3′ Reverse: 5′-GTGAGATGTAAAGGTATCCGAACCACACG-3′ |
5′-ACAGAAGCCGTAATCTATGTTGTCTATCGQTFCCTCCGGGAACTTA-3′ |
16 DNA molecules/μL |
Yes |
[46] |
This entry is adapted from the peer-reviewed paper 10.3390/bioengineering9100571
References
- Von Magnus, P.; Andersen, E.K.; Petersen, K.B.; Birch-Andersen, A. A pox-like disease in cynomolgus monkeys. Acta Pathol. Microbiol. Scand. 2009, 46, 156–176.
- Rondle, C.J.; Sayeed, K.A. Studies on monkeypox virus. Bull World Health Organ 1972, 46, 577–583.
- Prier, J.E.; Sauer, R.M. A pox disease of monkeys. Ann. N. Y. Acad. Sci. 1960, 85, 951–959.
- Zhu, H.; Fohlerová, Z.; Pekárek, J.; Basova, E.; Neužil, P. Recent advances in lab-on-a-chip technologies for viral diagnosis. Biosens. Bioelectron. 2020, 153, 112041.
- Artika, I.M.; Ma’Roef, C.N. Laboratory biosafety for handling emerging viruses. Asian Pac. J. Trop. Biomed. 2017, 7, 483–491.
- Jayakeerthi, R.S.; Potula, R.V.; Srinivasan, S.; Badrinath, S. Shell vial culture assay for the rapid diagnosis of Japanese encephalitis, west Nile and dengue-2 viral encephalitis. Virol. J. 2006, 3, 2.
- Battineni, G.; Chintalapudi, N.; Amenta, F. AI chatbot design during an epidemic like the novel coronavirus. Healthcare 2020, 8, 20154.
- Ahsan, M.M.; Uddin, M.R.; Luna, S.A. Monkeypox image data collection. arXiv 2022, arXiv:2206.01774.
- Ahsan, M.M.; Uddin, M.R.; Farjana, M.; Sakib, A.N.; Momin, K.; Luna, S.A. Image data collection and implementation of deep learning-based model in detecting monkeypox disease using modified VGG16. arXiv 2022, arXiv:2206.01862.
- Portela-Dias, J.; Sereno, S.; Falcão-Reis, I.; Rasteiro, C. Female monkeypox infection with localized genital lesions. Am. J. Obstet. Gynecol. 2022.
- Mailhe, M.; Beaumont, A.-L.; Thy, M.; Le Pluart, D.; Perrineau, S.; Houhou-Fidouh, N.; Deconinck, L.; Bertin, C.; Ferré, V.M.; Cortier, M.; et al. Clinical characteristics of ambulatory and hospitalized patients with monkeypox virus infection: An observational cohort study. Clin. Microbiol. Infect. 2022.
- Hierholzer, J.C.; Suggs, M.T.; Hall, E.C. Standardized viral hemagglutination and hemagglutination-inhibition tests II. Description and statistical evaluation. Appl. Microbiol. 1969, 18, 824–833.
- Sadeghi, P.; Sohrabi, H.; Hejazi, M.; Jahanban-Esfahlan, A.; Baradaran, B.; Tohidast, M.; Majidi, M.R.; Mokhtarzadeh, A.; Tavangar, S.M.; de la Guardia, M. Lateral flow assays (LFA) as an alternative medical diagnosis method for detection of virus species: The intertwine of nanotechnology with sensing strategies. TrAC Trends Anal. Chem. 2021, 145, 116460.
- Townsend, M.B.; MacNeil, A.; Reynolds, M.G.; Hughes, C.M.; Olson, V.A.; Damon, I.K.; Karem, K.L. Evaluation of the tetracore orthopox biothreat® antigen detection assay using laboratory grown orthopoxviruses and rash illness clinical specimens. J. Virol. Methods 2013, 187, 37–42.
- Stern, D.; Pauly, D.; Zydek, M.; Miller, L.; Piesker, J.; Laue, M.; Lisdat, F.; Dorner, M.B.; Dorner, B.G.; Nitsche, A. Development of a genus-specific antigen capture ELISA for orthopoxviruses—Target selection and optimized screening. PLoS ONE 2016, 11, e0150110.
- Ulaeto, D.O.; Lonsdale, S.G.; Laidlaw, S.M.; Clark, G.C.; Horby, P.; Carroll, M.W. A prototype lateral flow assay for detection of orthopoxviruses. Lancet Infect. Dis. 2022, 22, 1279–1280.
- Hughes, L.; Wilkins, K.; Goldsmith, C.S.; Smith, S.; Hudson, P.; Patel, N.; Karem, K.; Damon, I.; Li, Y.; Olson, V.A.; et al. A rapid orthopoxvirus purification protocol suitable for high-containment laboratories. J. Virol. Methods 2017, 243, 68–73.
- Richert-Pöggeler, K.R.; Franzke, K.; Hipp, K.; Kleespies, R.G. Electron microscopy methods for virus diagnosis and high resolution analysis of viruses. Front. Microbiol. 2019, 9, 3255.
- Lavazza, A.; Tittarelli, C.; Cerioli, M. The use of convalescent sera in immune-electron microscopy to detect non-suspected/new viral agents. Viruses 2015, 7, 2683–2703.
- Gelderblom, H.R.; Madeley, D. Rapid viral diagnosis of orthopoxviruses by electron microscopy: Optional or a must? Viruses 2018, 10, 142.
- Dumont, C.; Irenge, L.M.; Magazani, E.K.; Garin, D.; Muyembe, J.-J.T.; Bentahir, M.; Gala, J.-L. Simple technique for in field samples collection in the cases of skin rash illness and subsequent PCR detection of orthopoxviruses and varicella zoster virus. PLoS ONE 2014, 9, e96930.
- NCBI Monkeypox Genome Sequences. Available online: https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide&VirusLineage_ss=Monkeypoxvirus,taxid:10244&CreateDate_dt=2022-04-01T00:00:00.00ZTO2022-12-31T23:59:59.00Z (accessed on 9 August 2022).
- WHO Laboratory Testing for the Monkeypox Virus. Available online: https://www.who.int/publications/i/item/WHO-MPX-laboratory-2022.1 (accessed on 16 June 2022).
- Saxena, S.K.; Ansari, S.; Maurya, V.K.; Kumar, S.; Jain, A.; Paweska, J.T.; Tripathi, A.K.; Abdel-Moneim, A.S. Re-emerging human monkeypox: A major public-health debacle. J. Med. Virol. 2022.
- Ropp, S.L.; Jin, Q.; Knight, J.C.; Massung, R.F.; Esposito, J.J. PCR strategy for identification and differentiation of small pox and other orthopoxviruses. J. Clin. Microbiol. 1995, 33, 2069–2076.
- Meyer, H.; Ropp, S.L.; Esposito, J.J. Gene for a-type inclusion body protein is useful for a polymerase chain reaction assay to differentiate orthopoxviruses. J. Virol. Methods 1997, 64, 217–221.
- Meyer, H.; Damon, I.K.; Esposito, J.J. Orthopoxvirus diagnostics. In Vaccinia Virus and Poxvirology; Humana Press: Totowa, NJ, USA, 2004; pp. 119–133.
- Neubauer, H.; Reischl, U.; Ropp, S.; Esposito, J.; Wolf, H.; Meyer, H. Specific detection of monkeypox virus by polymerase chain reaction. J. Virol. Methods 1998, 74, 201–207.
- Li, Y.; Olson, V.A.; Laue, T.; Laker, M.T.; Damon, I.K. Detection of monkeypox virus with real-time PCR assays. J. Clin. Virol. 2006, 36, 194–203.
- Afonina, I.; Reed, M.; Lusby, E.; Shishkina, I.; Belousov, Y. Minor groove binder-conjugated DNA probes for quantitative DNA detection by hybridization-triggered fluorescence. BioTechniques 2002, 32, 940–949.
- Gelaye, E.; Mach, L.; Kolodziejek, J.; Grabherr, R.; Loitsch, A.; Achenbach, J.E.; Nowotny, N.; Diallo, A.; Lamien, C.E. A novel HRM assay for the simultaneous detection and differentiation of eight poxviruses of medical and veterinary importance. Sci. Rep. 2017, 7, srep42892.
- Nitsche, A.; Ellerbrok, H.; Pauli, G. Detection of orthopoxvirus DNA by real-time PCR and identification of variola virus DNA by melting analysis. J. Clin. Microbiol. 2004, 42, 1207–1213.
- Olson, V.A.; Laue, T.; Laker, M.T.; Babkin, I.V.; Drosten, C.; Shchelkunov, S.; Niedrig, M.; Damon, I.K.; Meyer, H. Real-time PCR system for detection of orthopoxviruses and simultaneous identification of smallpox virus. J. Clin. Microbiol. 2004, 42, 1940–1946.
- Simpson, K.; Heymann, D.; Brown, C.S.; Edmunds, W.J.; Elsgaard, J.; Fine, P.; Hochrein, H.; Hoff, N.A.; Green, A.; Ihekweazu, C.; et al. Human monkeypox—After 40 years, an unintended consequence of smallpox eradication. Vaccine 2020, 38, 5077–5081.
- Hughes, C.M.; Liu, L.; Davidson, W.B.; Radford, K.W.; Wilkins, K.; Monroe, B.; Metcalfe, M.G.; Likafi, T.; Lushima, R.S.; Kabamba, J.; et al. A tale of two viruses: Coinfections of monkeypox and varicella zoster virus in the Democratic Republic of Congo. Am. J. Trop. Med. Hyg. 2021, 104, 604–611.
- Luciani, L.; Inchauste, L.; Ferraris, O.; Charrel, R.; Nougairède, A.; Piorkowski, G.; Peyrefitte, C.; Bertagnoli, S.; de Lamballerie, X.; Priet, S. A novel and sensitive real-time PCR system for universal detection of poxviruses. Sci. Rep. 2021, 11, 1798.
- Maksyutov, R.A.; Gavrilova, E.V.; Shchelkunov, S.N. Species-specific differentiation of variola, monkeypox, and varicella-zoster viruses by multiplex real-time PCR assay. J. Virol. Methods 2016, 236, 215–220.
- Li, Y.; Meyer, H.; Zhao, H.; Damon, I.K. GC Content-based pan-pox universal PCR assays for poxvirus detection. J. Clin. Microbiol. 2010, 48, 268–276.
- Glökler, J.; Lim, T.S.; Ida, J.; Frohme, M. Isothermal amplifications—A comprehensive review on current methods. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 543–586.
- Becherer, L.; Borst, N.; Bakheit, M.; Frischmann, S.; Zengerle, R.; von Stetten, F. Loop-mediated isothermal amplification (LAMP)—Review and classification of methods for sequence-specific detection. Anal. Methods 2020, 12, 717–746.
- Nagamine, K.; Hase, T.; Notomi, T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell Probes 2002, 16, 223–229.
- Iizuka, I.; Saijo, M.; Shiota, T.; Ami, Y.; Suzaki, Y.; Nagata, N.; Hasegawa, H.; Sakai, K.; Fukushi, S.; Mizutani, T.; et al. Loop-mediated isothermal amplification-based diagnostic assay for monkeypox virus infections. J. Med. Virol. 2009, 81, 1102–1108.
- Bhadra, S.; Ellington, A.D. Portable nucleic acid tests for rapid detection of monkeypox virus. medRxiv 2022.
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. TrAC Trends Anal. Chem. 2017, 98, 19–35.
- Fan, X.; Li, L.; Zhao, Y.; Liu, Y.; Liu, C.; Wang, Q.; Dong, Y.; Wang, S.; Chi, T.; Song, F.; et al. Clinical validation of two recombinase-based isothermal amplification assays (RPA/RAA) for the rapid detection of African swine fever virus. Front. Microbiol. 2020, 11, 1696.
- Davi, S.D.; Kissenkötter, J.; Faye, M.; Böhlken-Fascher, S.; Stahl-Hennig, C.; Faye, O.; Faye, O.; Sall, A.A.; Weidmann, M.; Ademowo, O.G.; et al. Recombinase polymerase amplification assay for rapid detection of monkeypox virus. Diagn. Microbiol. Infect. Dis. 2019, 95, 41–45.
- Li, Y.; Zhao, H.; Wilkins, K.; Hughes, C.; Damon, I.K. Real-time PCR assays for the specific detection of monkeypox virus West African and Congo Basin strain DNA. J. Virol. Methods 2010, 169, 223–227.
- Kulesh, D.A.; Loveless, B.M.; Norwood, D.; Garrison, J.; Whitehouse, C.A.; Hartmann, C.; Mucker, E.; Miller, D.; Wasieloski, L.P.; Huggins, J.; et al. Monkeypox virus detection in rodents using real-time 3′-minor groove binder TaqMan® assays on the roche light cycler. Lab. Investig. 2004, 84, 1200–1208.
- Shchelkunov, S.N.; Shcherbakov, D.N.; Maksyutov, R.A.; Gavrilova, E.V. Species-specific identification of variola, monkeypox, cowpox, and vaccinia viruses by multiplex real-time PCR assay. J. Virol. Methods 2011, 175, 163–169.