Organogold chemistry is the study of compounds containing gold–carbon bonds. They are studied in academic research, but have not received widespread use otherwise. The dominant oxidation states for organogold compounds are I with coordination number 2 and a linear molecular geometry and III with CN = 4 and a square planar molecular geometry. The first organogold compound discovered was gold(I) carbide Au2C2, which was first prepared in 1900.
- molecular geometry
- organogold
- square planar
1. Gold(I)
Gold(I) complexes are 2-coordinate, linear, diamagnetic, 14 electron species.[1][2][3] They typically exist as adducts LAuR with as ligand L for instance a triphenylphosphine or an isocyanide. The ligand prevents reduction of Au(I) to metallic Au(0) with dimerization of the organic residue. Gold(I) can also exist as the aurate M[AuR2] (the ate complex) whereby the cation is usually fitted with a complexing agent to improve stability. The AuR2− anion is also linear just as other M(d10) species such as Hg(Me)2 and Pd(Me)22+. Gold is known to form acetylides (capable of forming polymeric structures), carbenes and carbynes. The classic method for the preparation of LAuR compounds is by reaction of a Grignard reagent with a gold(I) halide. A subsequent reaction with an organolithium R-Li forms the ate complex.
In a special group of compounds, an aryl carbon atom acts as a bridge between two gold atoms. One such compound, (MesAu)5, is formed in a reaction between Au(CO)Cl and the mesityl Grignard. Carbon can be coordinated with gold up to a value to 6. Compounds of the type C(AuL)4 are isolobal with methane and those of type C(AuL)5+ isolobal with the methanium ion. These hypercoordinated organogold clusters are often stabilized by aurophilic interactions between the formally closed-shell gold centers.[4]
-
Some typical organogold species with assorted bonding modes. https://handwiki.org/wiki/index.php?curid=1641699
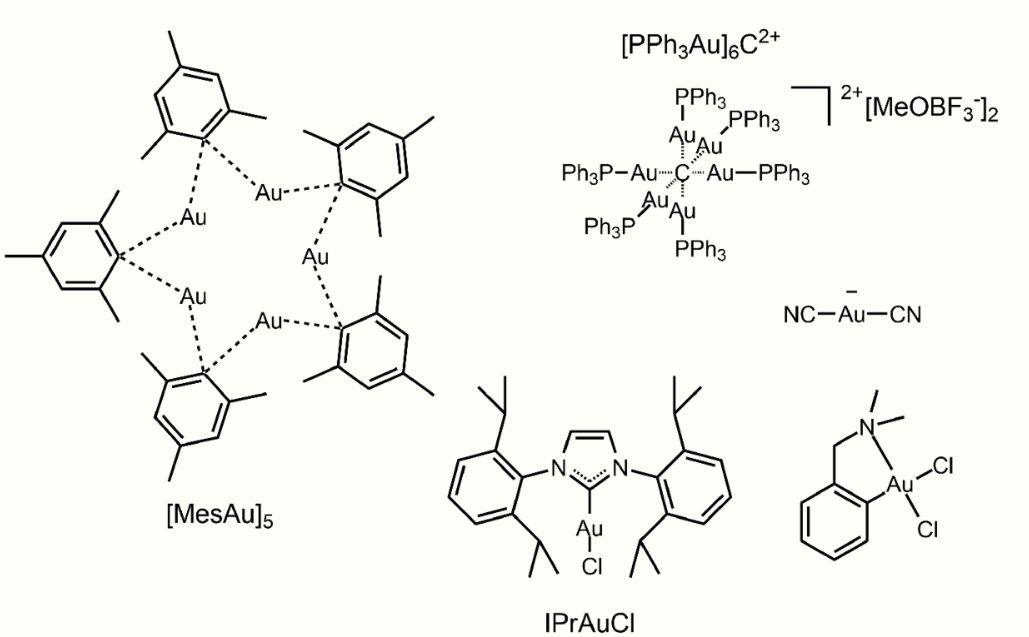
Gold cyanide compounds (MAu(CN)2) are of some importance to gold cyanidation, a process for the extraction of gold from low-grade ore. The carbon to metal bond in metal cyanides is usually ionic but evidence exists that the C-Au bonding in the gold cyanide ion is covalent.[5]
2. Gold(III)
Gold(III) complexes are 4 coordinate, square planar, diamagnetic, toxic, 16 electron species. When the formal coordination number is less than 4, ligands such as chlorine can make up for it by forming a bridging ligand. Intramolecular chelation is another strategy. In general gold(III) compounds are toxic and therefore less studied than gold(I). Monoarylgold(III) complexes are one well-studied class of complexes. They are often prepared by direct electrophilic auration of arenes by AuCl3.[6] Homoleptic tetraalkylaurate(III) complexes (e.g. Li[AuMe4]) are also well-characterized.[7]
3. Gold Catalysis
3.1. General Considerations
Gold-catalyzed reactions fall into two major categories: heterogeneous catalysis including catalysts by gold nanoparticles (e.g., Au/TiO2) and thiol-monolayer gold surfaces, and catalysts on alumina support, including alumina supported Au/CeO2. These catalysts have been investigated for industrially important processes like the oxidation of alcohols, oxidation of carbon monoxide (CO), and various selective hydrogenation reactions (e.g. butadiene to butene). Though often efficient and exhibiting useful or unique selectivities, there is considerable uncertainty with respect to the mechanism of processes catalyzed by various heterogeneous gold catalysts, even compared to other heterogeneous transition metal catalysts.
In contrast, homogeneous catalysis with gold uses simple or ligand-bound gold(I) or gold(III) compounds that are soluble in organic solvents and is used for the synthesis of fine chemicals in organic chemistry.[8][9] Binary gold halides and simple complexes, including gold(I) chloride, gold(III) chloride, and chloroauric acid, have been employed as complexes. These gold sources, however, quickly give rise to ill-defined and easily deactivated (via reduction to Au0) active catalysts in solution. The development of well-defined phosphine- or NHC-ligated gold(I) complexes was an important advance and led to significant increase in interest in the synthetic applications of gold catalysis. Ligated gold(I) complexes are typically prepared and stored as the bench-stable (but unreactive) chlorides, LAuCl, e.g., chloro(triphenylphosphine)gold(I), which are typically activated via halide abstraction with silver salts like AgOTf, AgBF4, or AgSbF6 to generate a cationic gold(I) species.[10][11] Although the coordinatively unsaturated complex "LAu+" is notionally generated from a LAuCl/AgX mixture, the exact nature of the cationic gold species and the role of the silver salt remains somewhat contentious.[12][13][14] The para-nitrobenzoate, bistriflimide, and certain nitrile complexes represent catalytically active yet isolable silver-free precatalysts.
Cationic gold(I) forms π-complexes with alkene or alkyne bonds, following the Dewar–Chatt–Duncanson model. Gold is certainly not the only metal showing this type of bonding and reactivity, several metal ions isolobal with the simple proton (i.e., an empty s-orbital) do as well: for example, mercury(II) and platinum(II). Electrophilic ions and complexes such as these with a strong propensity to form π-complexes are generally known as pi(π)-acids (see also: cation–pi interaction).[15]
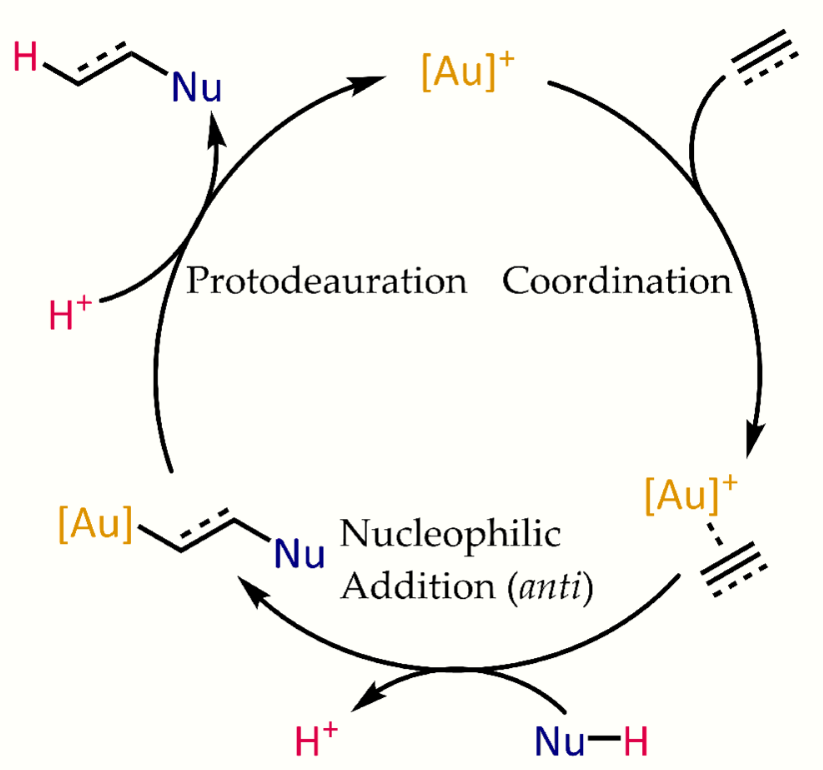
Gold(I)-alkene and -alkyne complexes are electrophilic and susceptible toward nucleophilic attack. In oxymercuration the resultant organomercurial species is generated stoichiometrically, and requires an additional step to liberate the product. In the case of gold, protonolysis of the Au-C bond closes the catalytic cycle, allowing the coordination of another substrate. Some practical advantages of gold(I) catalysis include: 1) air stability (due to the high oxidation potential of Au(I)), 2) tolerance towards adventitious moisture (due its low oxophilicity), and 3) relatively low toxicity compared to other pi-acids (e.g., Pt(II) and Hg(II)). Chemically, Au(I) complexes typically do not undergo oxidation to higher oxidation states, and Au(I)-alkyls and -vinyls are not susceptible to β hydride elimination.[16]
-
Typical mechanism for the gold(I)-catalyzed hydrofunctionalization of alkynes and allenes. https://handwiki.org/wiki/index.php?curid=1671562
3.2. Historical Development
In 1976, Thomas and coworkers reported conversion of phenylacetylene to acetophenone using tetrachloroauric acid in a 37% yield.[17] In this reaction gold(III) was used as a homogeneous catalyst replacing mercury in oxymercuration. This same study lists a published yield >150%, indicating catalysis that perhaps was not acknowledged by the chemists.
In 1991, Utimoto reacted gold(III) (NaAuCl4) with alkynes and water.[18] Teles identified a major drawback of this method as Au(III) was rapidly reduced to catalytically dead metallic gold and in 1998 returned to the theme of ligand supported Au(I) for the same transformation:[19]
-
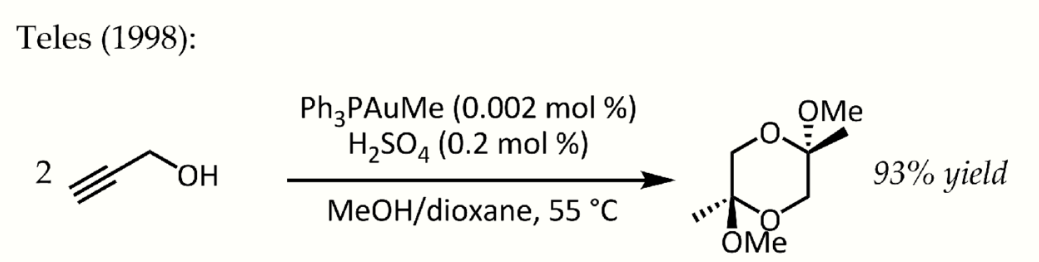 Teles gold catalysis. https://handwiki.org/wiki/index.php?curid=1610425
Teles gold catalysis. https://handwiki.org/wiki/index.php?curid=1610425
This particular reaction demonstrated fantastic catalytic efficiency and would trigger a flurry of research into the use of phosphinegold(I) complexes for the activation C-C multiple bonds in the years to come.[20] In spite of the lower stability of gold(III) complexes under catalytic conditions, simple AuCl3 was also found to be an efficient catalyst in some cases. For instance, Hashmi reported an AuCl3-catalyzed alkyne / furan Diels–Alder reaction - a type of cycloaddition that does not ordinarily occur - for the synthesis of 2,3-disubstituted phenols:[21]
-
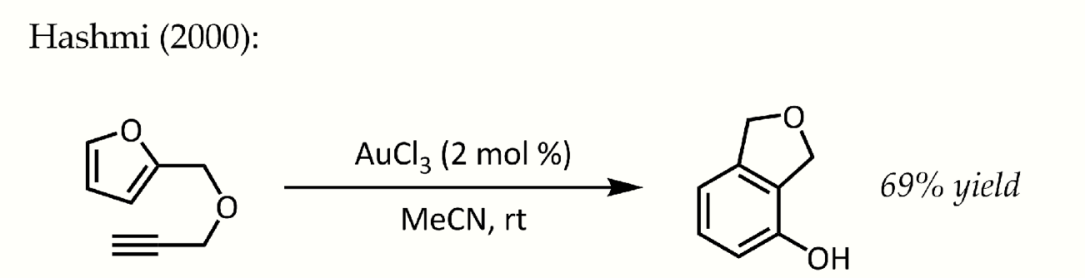 Hashmi phenol synthesis. https://handwiki.org/wiki/index.php?curid=1922634
Hashmi phenol synthesis. https://handwiki.org/wiki/index.php?curid=1922634
Further mechanistic studies conclude that this is not a concerted transformation, but rather an initial alkyne hydroarylation followed by a series of non-obvious intramolecular rearrangements, concluding with a 6π electrocyclization and rearomatization.
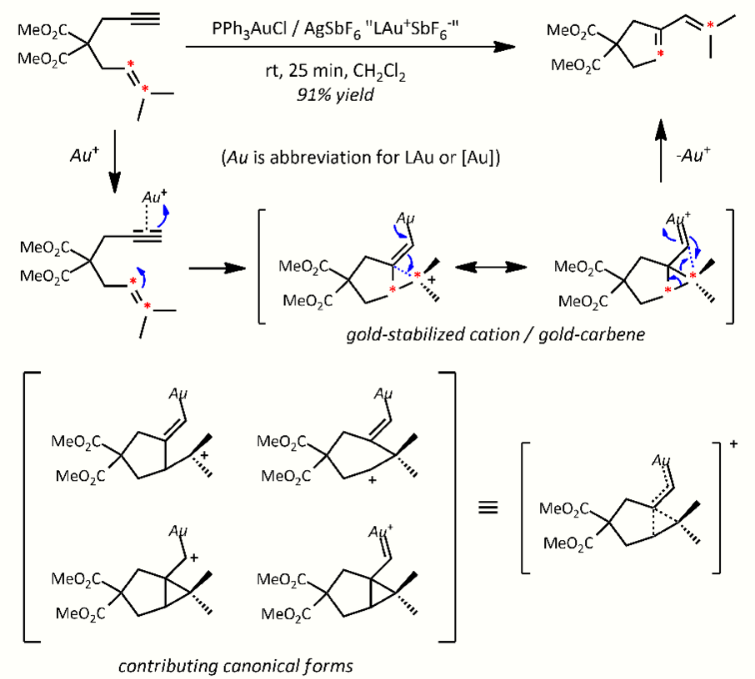
Relativistic effects are significant in organogold chemistry due to the large nuclear charge of the metal (Z = 79). As a consequence of relativistically expanded 5d orbitals, the LAu fragment can stabilize a neighboring carbocation via electron donation into the empty p-type orbital. Thus, in addition to their expected carbocation-like reactivity, these cations also exhibit significant carbene character, a property that has been exploited in catalytic transformations such as cyclopropanation and C-H insertion.[22] Propargyl esters can serve as precursors for cationic gold-vinylcarbene intermediates, which can react with alkenes in a concerted manner to afford the cyclopropanation product. The use of a chiral ligand ((R)-DTBM-SEGPHOS) resulted in good to excellent levels of enantioselectivity.[23]
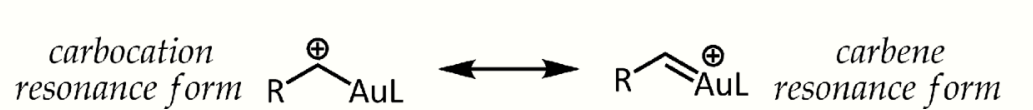 Alpha-gold cations. https://handwiki.org/wiki/index.php?curid=1429682
Alpha-gold cations. https://handwiki.org/wiki/index.php?curid=1429682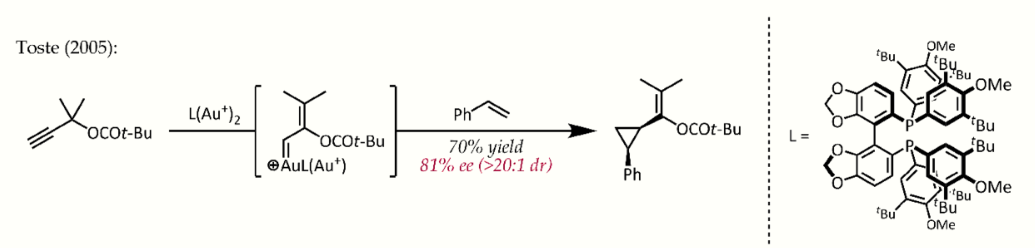
Although Echavarren first reported the preparation of chiral bisphosphinedigold(I) complexes for enantioselective gold catalysis proceeding via the typical pi-activation mechanism,[24] an early, atypical example of enantioselective catalysis by gold was described by Hayashi and Ito in 1986.[25] In this process, benzaldehyde and methyl isocyanoacetate undergo cyclization in the presence of a chiral ferrocenylphosphine ligand and a bis(isocyanide)gold(I) complex to form a chiral oxazoline. Since oxazolines can be hydrolyzed to provide a 1,2-aminoalcohol, this reaction constitutes the first example of a catalytic, asymmetric aldol reaction.
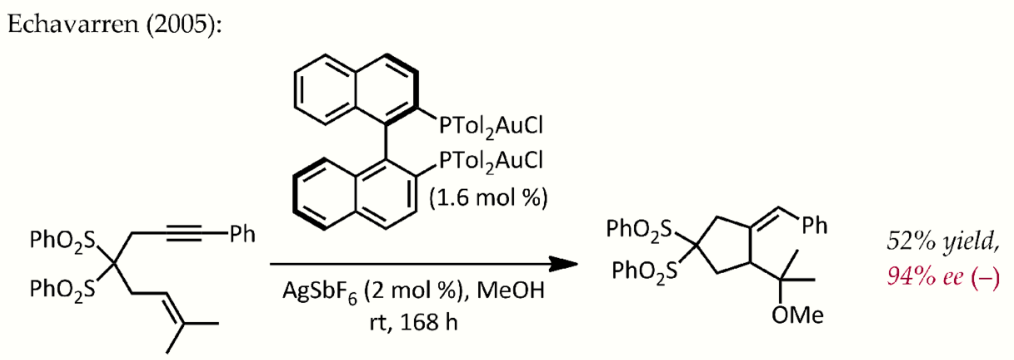
Echavarren gold phosphine enantioselective. https://handwiki.org/wiki/index.php?curid=1270311
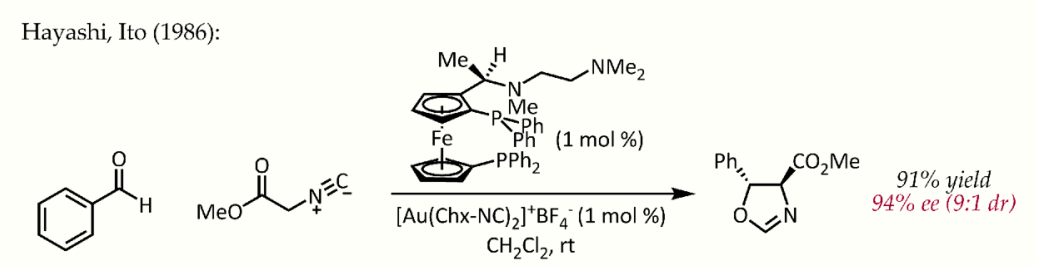
Hayashi ito aldol. https://handwiki.org/wiki/index.php?curid=1100964
In contrast to the other reactions described above, this reaction does not involve activation of a C-C double or triple bond by gold. In a simple mechanistic picture, gold(I) simultaneously coordinates to two phosphine ligands and the carbon isocyanate group [26] which is then attacked by the carbonyl group. Further studies on the bonding mode of Au(I) indicate that this simple picture may have to be revised.
Heterogeneous gold catalysis is an older science. Gold is an attractive metal to use because of its stability against oxidation and its variety in morphology for instance gold cluster materials. Gold has been shown to be effective in low-temperature CO oxidation and acetylene hydrochlorination to vinyl chlorides. The exact nature of the catalytic site in this type of process is debated.[27] The notion that gold can catalyse a reaction does not imply it is the only way. However, other metals can do the same job inexpensively, notably in recent years iron (see organoiron chemistry).
4. Gold Catalyzed Reactions
Although of no commercial importance, gold catalyzes many organic transformations, usually carbon-carbon bond formation from Au(I), and C-X (X = O, N) bond formation from the Au(III) state, due to that ion's harder Lewis acidity. During the past decade, several studies have demonstrated that gold can efficiently catalyze C-C and C-heteroatom cross-coupling reactions that proceed through an Au(I)/Au(III) cycle.[28] Hong C. Shen summarized homogeneous reactions forming cyclic compounds into 4 main categories:[29]
- heteroatom nucleophilic addition to unsaturated C-C bonds, especially to form small heterocycles (furans, pyrroles, thiophenes)
- Hydroarylation: basically a Friedel-Crafts reaction using metal-alkyne complexes. Example, the reaction of mesitylene with phenylacetylene:[30]
-
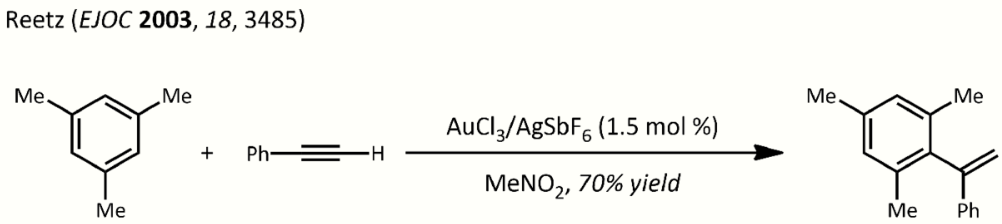 Hydroarylation reetz. https://handwiki.org/wiki/index.php?curid=1559468
Hydroarylation reetz. https://handwiki.org/wiki/index.php?curid=1559468
- Enyne cyclization, in particular cycloisomerization, one early example being a 5-exo-dig 1,6 enyne cycloisomerization:[31]
-
 1,6-enyne mechanism. https://handwiki.org/wiki/index.php?curid=1922933
1,6-enyne mechanism. https://handwiki.org/wiki/index.php?curid=1922933
- cycloaddition reactions with early example the cycloaddition of a nitrile oxide with an alkyne.[32]
Other reactions are the use of gold in C–H bond activation[33] and aldol reactions. Gold also catalyses coupling reactions.[34]
4.1. Limitations
While gold-catalyzed hydrofunctionalization of alkynes, allenes, and allylic alcohols[35] occurs readily under comparatively mild conditions, unactivated alkene remain poor substrates in most cases,[36] in large part due to the resistance of the intermediate alkylgold(I) complexes to protodeauration.[37] The development of intermolecular gold-catalyzed transformations has also lagged behind the development of intramolecular ones.[38]
The content is sourced from: https://handwiki.org/wiki/Chemistry:Organogold_chemistry
References
- Elschenbroich, C. and Salzer, A. (1992) Organometallics : A Concise Introduction. Wiley-VCH: Weinheim. ISBN:3-527-28165-7
- Parish, R. V. (1997). "Organogold chemistry: II reactions". Gold Bulletin 30 (2): 55–62. doi:10.1007/BF03214757. https://dx.doi.org/10.1007%2FBF03214757
- Parish, R. V. (1998). "Organogold chemistry: III applications". Gold Bulletin 31: 14–21. doi:10.1007/BF03215470. https://dx.doi.org/10.1007%2FBF03215470
- Schmidbaur, Hubert; Schier, Annette (2011-12-05). "Aurophilic interactions as a subject of current research: an up-date" (in en). Chemical Society Reviews 41 (1): 370–412. doi:10.1039/C1CS15182G. ISSN 1460-4744. PMID 21863191. https://dx.doi.org/10.1039%2FC1CS15182G
- Wang, X. B.; Wang, Y. L.; Yang, J.; Xing, X. P.; Li, J.; Wang, L. S. (2009). "Evidence of Significant Covalent Bonding in Au(CN)2−". Journal of the American Chemical Society 131 (45): 16368–70. doi:10.1021/ja908106e. PMID 19860420. https://dx.doi.org/10.1021%2Fja908106e
- Kharasch, M. S.; Isbell, Horace S. (1931-08-01). "The Chemistry of Organic Gold Compounds. III. Direct Introduction of Gold into the Aromatic Nucleus (Preliminary Communication)". Journal of the American Chemical Society 53 (8): 3053–3059. doi:10.1021/ja01359a030. ISSN 0002-7863. https://dx.doi.org/10.1021%2Fja01359a030
- Rice, Gary W.; Tobias, R. Stuart. (1975-10-01). "Synthesis of tetramethylaurate(III). Structures of lithium dimethylaurate and lithium tetramethylaurate in solution". Inorganic Chemistry 14 (10): 2402–2407. doi:10.1021/ic50152a020. ISSN 0020-1669. https://dx.doi.org/10.1021%2Fic50152a020
- Gold catalysis for organic synthesis F. Dean Toste (Editor) Thematic Series in the Open Access Beilstein Journal of Organic Chemistry http://www.beilstein-journals.org/bjoc/browse/singleSeries.htm?sn=15
- Raubenheimer, H. G.; Schmidbaur, H. (2014). "The Late Start and Amazing Upswing in Gold Chemistry". Journal of Chemical Education 91 (12): 2024–2036. doi:10.1021/ed400782p. Bibcode: 2014JChEd..91.2024R. https://dx.doi.org/10.1021%2Fed400782p
- Ranieri, Beatrice; Escofet, Imma; Echavarren, Antonio M. (2015-06-24). "Anatomy of gold catalysts: facts and myths" (in en). Org. Biomol. Chem. 13 (26): 7103–7118. doi:10.1039/c5ob00736d. ISSN 1477-0539. PMID 26055272. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4479959
- Wang, Yi-Ming; Lackner, Aaron D.; Toste, F. Dean (2013-11-14). "Development of Catalysts and Ligands for Enantioselective Gold Catalysis" (in EN). Accounts of Chemical Research 47 (3): 889–901. doi:10.1021/ar400188g. PMID 24228794. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3960333
- Zhdanko, Alexander; Maier, Martin E. (2015-09-09). "Explanation of "Silver Effects" in Gold(I)-Catalyzed Hydroalkoxylation of Alkynes" (in EN). ACS Catalysis 5 (10): 5994–6004. doi:10.1021/acscatal.5b01493. https://dx.doi.org/10.1021%2Facscatal.5b01493
- Homs, Anna; Escofet, Imma; Echavarren, Antonio M. (2013). "On the Silver Effect and the Formation of Chloride-Bridged Digold Complexes". Organic Letters 15 (22): 5782–5785. doi:10.1021/ol402825v. PMID 24195441. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3833279
- Wang, Dawei; Cai, Rong; Sharma, Sripadh; Jirak, James; Thummanapelli, Sravan K.; Akhmedov, Novruz G.; Zhang, Hui; Liu, Xingbo et al. (2012-05-18). ""Silver Effect" in Gold(I) Catalysis: An Overlooked Important Factor" (in EN). Journal of the American Chemical Society 134 (21): 9012–9019. doi:10.1021/ja303862z. PMID 22563621. https://dx.doi.org/10.1021%2Fja303862z
- Fürstner, A.; Davies, P. W. (2007). "Catalytic Carbophilic Activation: Catalysis by Platinum and Gold π Acids". Angewandte Chemie International Edition 46 (19): 3410–3449. doi:10.1002/anie.200604335. PMID 17427893. https://dx.doi.org/10.1002%2Fanie.200604335
- Shen, H. C. (2008). "Recent advances in syntheses of heterocycles and carbocycles via homogeneous gold catalysis. Part 1: Heteroatom addition and hydroarylation reactions of alkynes, allenes, and alkenes". Tetrahedron 64 (18): 3885–3903. doi:10.1016/j.tet.2008.01.081. https://dx.doi.org/10.1016%2Fj.tet.2008.01.081
- Norman, R. O. C.; Parr, W. J. E.; Thomas, C. B. (1976). "The reactions of alkynes, cyclopropanes, and benzene derivatives with gold(III)". Journal of the Chemical Society, Perkin Transactions 1 (18): 1983. doi:10.1039/P19760001983. https://dx.doi.org/10.1039%2FP19760001983
- Fukuda, Y.; Utimoto, K. (1991). "Effective transformation of unactivated alkynes into ketones or acetals with a gold(III) catalyst". The Journal of Organic Chemistry 56 (11): 3729–3731. doi:10.1021/jo00011a058. https://dx.doi.org/10.1021%2Fjo00011a058
- 3.0.CO;2-N. PMID 29710887. https://dx.doi.org/10.1002%2F%28SICI%291521-3773%2819980605%2937%3A10%3C1415%3A%3AAID-ANIE1415%3E3.0.CO%3B2-N" id="ref_19">Teles, J. H.; Brode, S.; Chabanas, M. (1998). "Cationic Gold(I) Complexes: Highly Efficient Catalysts for the Addition of Alcohols to Alkynes". Angewandte Chemie International Edition 37 (10): 1415–1418. doi:10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.0.CO;2-N. PMID 29710887. https://dx.doi.org/10.1002%2F%28SICI%291521-3773%2819980605%2937%3A10%3C1415%3A%3AAID-ANIE1415%3E3.0.CO%3B2-N
- Nugent, W. A. (2012). ""Black Swan Events" in Organic Synthesis". Angewandte Chemie International Edition 51 (36): 8936–49. doi:10.1002/anie.201202348. PMID 22893229. https://dx.doi.org/10.1002%2Fanie.201202348
- Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. (2000). "Highly Selective Gold-Catalyzed Arene Synthesis". Journal of the American Chemical Society 122 (46): 11553–11554. doi:10.1021/ja005570d. https://dx.doi.org/10.1021%2Fja005570d
- Gorin, David J.; Toste, F. Dean (2007). "Relativistic effects in homogeneous gold catalysis". Nature 446 (7134): 395–403. doi:10.1038/nature05592. PMID 17377576. Bibcode: 2007Natur.446..395G. https://dx.doi.org/10.1038%2Fnature05592
- Johansson, Magnus J.; Gorin, David J.; Staben, Steven T.; Toste, F. Dean (2005-11-30). "Gold(I)-Catalyzed Stereoselective Olefin Cyclopropanation" (in en). Journal of the American Chemical Society 127 (51): 18002–18003. doi:10.1021/ja0552500. PMID 16366541. https://dx.doi.org/10.1021%2Fja0552500
- Muñoz, M. Paz; Adrio, Javier; Carretero, Juan Carlos; Echavarren, Antonio M. (2005-02-12). "Ligand Effects in Gold- and Platinum-Catalyzed Cyclization of Enynes: Chiral Gold Complexes for Enantioselective Alkoxycyclization" (in en). Organometallics 24 (6): 1293–1300. doi:10.1021/om0491645. https://dx.doi.org/10.1021%2Fom0491645
- Ito, Y.; Sawamura, M.; Hayashi, T. (1986). "Catalytic asymmetric aldol reaction: Reaction of aldehydes with isocyanoacetate catalyzed by a chiral ferrocenylphosphine-gold(I) complex". Journal of the American Chemical Society 108 (20): 6405–6406. doi:10.1021/ja00280a056. https://dx.doi.org/10.1021%2Fja00280a056
- Togni, A.; Pastor, S. D. (1990). "Chiral cooperativity: The nature of the diastereoselective and enantioselective step in the gold(I)-catalyzed aldol reaction utilizing chiral ferrocenylamine ligands". The Journal of Organic Chemistry 55 (5): 1649–1664. doi:10.1021/jo00292a046. https://dx.doi.org/10.1021%2Fjo00292a046
- Hutchings, G. J.; Brust, M.; Schmidbaur, H. (2008). "Gold—an introductory perspective". Chemical Society Reviews 37 (9): 1759–65. doi:10.1039/b810747p. PMID 18762825. https://dx.doi.org/10.1039%2Fb810747p
- Nijamudheen, A.; Datta, Ayan (2020). "Gold‐Catalyzed Cross‐Coupling Reactions: An Overview of Design Strategies, Mechanistic Studies, and Applications". Chemistry: A European Journal 26 (7): 1442–1487. doi:10.1002/chem.201903377. PMID 31657487. https://dx.doi.org/10.1002%2Fchem.201903377
- Shen, H. C. (2008). "Recent advances in syntheses of carbocycles and heterocycles via homogeneous gold catalysis. Part 2: Cyclizations and cycloadditions". Tetrahedron 64 (34): 7847–7870. doi:10.1016/j.tet.2008.05.082. https://dx.doi.org/10.1016%2Fj.tet.2008.05.082
- Reetz, M. T.; Sommer, K. (2003). "Gold-Catalyzed Hydroarylation of Alkynes". European Journal of Organic Chemistry 2003 (18): 3485–3496. doi:10.1002/ejoc.200300260. https://dx.doi.org/10.1002%2Fejoc.200300260
- Nieto-Oberhuber, C.; Muñoz, M. P.; Buñuel, E.; Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. (2004). "Cationic Gold(I) Complexes: Highly Alkynophilic Catalysts for theexo- andendo-Cyclization of Enynes". Angewandte Chemie International Edition 43 (18): 2402–2406. doi:10.1002/anie.200353207. PMID 15114573. https://dx.doi.org/10.1002%2Fanie.200353207
- Gasparrini, F.; Giovannoli, M.; Misiti, D.; Natile, G.; Palmieri, G.; Maresca, L. (1993). "Gold(III)-catalyzed one-pot synthesis of isoxazoles from terminal alkynes and nitric acid". Journal of the American Chemical Society 115 (10): 4401–4402. doi:10.1021/ja00063a084. https://dx.doi.org/10.1021%2Fja00063a084
- Hoffmann-Röder, A.; Krause, N. (2005). "The golden gate to catalysis". Organic & Biomolecular Chemistry 3 (3): 387–91. doi:10.1039/b416516k. PMID 15678171. https://dx.doi.org/10.1039%2Fb416516k
- Wegner, H. A.; Auzias, M. (2011). "Gold for C-C coupling reactions: a Swiss-Army-knife catalyst?". Angewandte Chemie International Edition 50 (36): 8236–47. doi:10.1002/anie.201101603. PMID 21818831. https://dx.doi.org/10.1002%2Fanie.201101603
- Bandini, Marco (2011-02-01). "Allylic Alcohols: Sustainable Sources for Catalytic Enantioselective Alkylation Reactions" (in en). Angewandte Chemie International Edition 50 (5): 994–995. doi:10.1002/anie.201006522. ISSN 1521-3773. PMID 21268189. https://dx.doi.org/10.1002%2Fanie.201006522
- Zhang, Zhibin; Lee, Seong Du; Widenhoefer, Ross A. (2009-04-22). "Intermolecular Hydroamination of Ethylene and 1-Alkenes with Cyclic Ureas Catalyzed by Achiral and Chiral Gold(I) Complexes". Journal of the American Chemical Society 131 (15): 5372–5373. doi:10.1021/ja9001162. ISSN 0002-7863. PMID 19326908. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2891684
- LaLonde, Rebecca L.; Jr., William E. Brenzovich; Benitez, Diego; Tkatchouk, Ekaterina; Kelley, Kotaro; III, William A. Goddard; Toste, F. Dean (2010). "Alkylgold complexes by the intramolecular aminoauration of unactivated alkenes" (in en). Chemical Science 1 (2): 226. doi:10.1039/C0SC00255K. PMID 24358445. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3866133
- Muratore, Michael E.; Homs, Anna; Obradors, Carla; Echavarren, Antonio M. (2014-11-01). "Meeting the Challenge of Intermolecular Gold(I)-Catalyzed Cycloadditions of Alkynes and Allenes" (in en). Chemistry: An Asian Journal 9 (11): 3066–3082. doi:10.1002/asia.201402395. ISSN 1861-471X. PMID 25048645. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4676923