Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Notably, 5% of metastatic colorectal cancer (mCRC) cases harbor Erb-B2 receptor tyrosine kinase 2 (ERBB2) alterations. ERBB2, commonly referred to as human epidermal growth factor receptor 2, is a member of the human epidermal growth factor receptor family of protein tyrosine kinases. In addition to being a recognized therapeutic target in the treatment of gastric and breast malignancies, it is considered crucial in the management of colorectal cancer (CRC).
- ERBB2
- HER2
- targeted therapy
- colorectal cancer
1. Structural Features of the ERBB2 Receptor
The four ERBB (or EGFR) proteins are members of subclass I of the RTK superfamily; they include EGFR/ERBB1/HER1, ERBB2/HER2, ERBB3/HER3, and ERBB4/HER4. These receptors are essential for a variety of cellular processes, including cell growth, proliferation, differentiation, and migration [1]. In addition to an extracellular domain (ECD) or ligand-binding area and a single transmembrane domain (TMD), all receptors comprise a cytoplasmic/intracellular region composed of a juxtamembrane domain (JMD), a kinase domain, and a C-terminal tail domain (Figure 1) [2][3]. The ECD comprises four subdomains (I-IV); in the absence of a ligand, domains II and IV adopt an auto-inhibited tethered (closed) conformation. Upon ligand interaction between domains I and III, the dimerization arm in domain II unhinges, leading to receptor homo- or heterodimerization, allosteric kinase activation, and C-terminal tail domain phosphorylation [3]. This process recruits and activates various downstream signaling proteins containing Src homologous structure-2 (SH2) or phosphotyrosine binding structural domains and engages downstream mediators to drive important cellular signaling pathways [4]. Despite comparable levels of total phosphotyrosine, EGF-activated Erb-B2 receptor tyrosine kinase 2 (ERBB2) binds Shc considerably weakly compared to ERBB2 that has been dimerized consequent to mutations in the transmembrane structural domain [5]. Therefore, the signal produced by receptor heterodimers is distinctive; instead of being the simple sum of individual dimer partner signaling proper ties, it reflects the properties of the heterodimers.
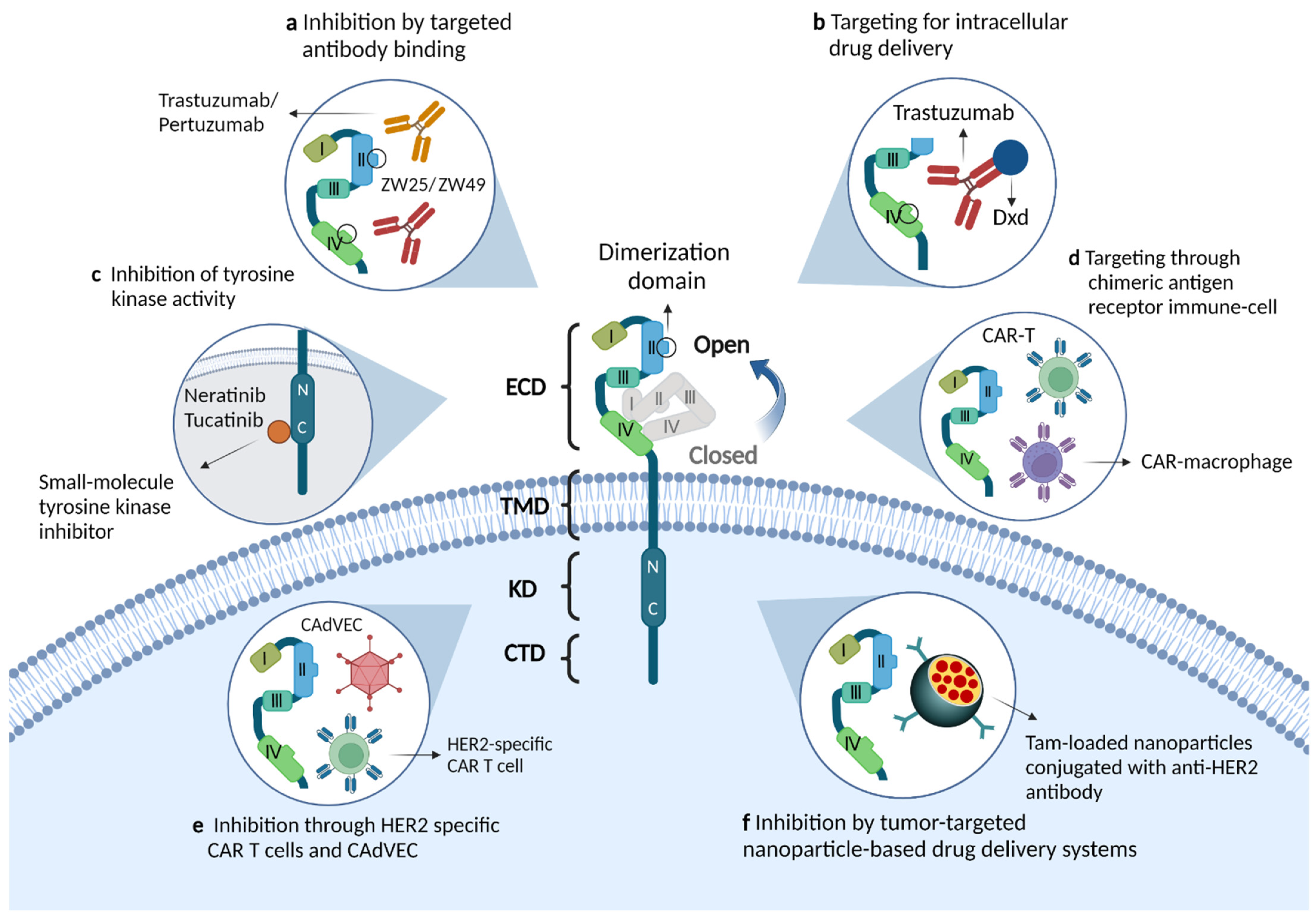
Figure 1. Molecular structure of the ERBB2/HER2 receptor and the mechanism of action of novel therapeutic agents. (a) Monoclonal antibodies that target binding to the extracellular structural domain of HER2, including single-epitope monoclonal antibodies (trastuzumab, pertuzumab) and bispecific HER2 antibodies (ZW25, ZW49). The antitumor effects of these drugs are mediated through a variety of mechanisms: inhibition of downstream signaling pathways, involvement in antibody-dependent cellular cytotoxicity or inhibition of receptor dimerization. (b) Antibody-drug conjugates targeting HER2, such as trastuzumab emtansine and trastuzumab deruxtecan, release and deliver cytotoxic drugs while binding antibodies and thus exert anti-tumor effects. (c) Small molecule inhibitors, such as neratinib and tucatinib, inhibit activation of the PI3K pathway by binding to the tyrosine-kinase domain of the HER2 receptor. (d,e) HER2-targeted immunotherapies, such as HER2-specific chimeric antigen receptor immune cells or modified oncolytic virus, enhance immune cell activity and stimulate anti-tumor immune responses. (f) Tumor-targeting nanoparticles, such as nanodelivery systems with internal encapsulation of tamoxifen and external loading of anti-HER2 antibodies, can efficiently kill HER2 overexpressing breast cancer cells by penetrating the tumor microenvironment.
Numerous ligands bind to the ECD of the ERBB receptor; these include EGF, neuregulin, and transforming growth factor-α, among others [6][7]. Growth factors that cause receptor dimerization and/or oligomerization upon binding to extracellular areas of the receptor represent receptor-specific ligands which typically activate ERBB family members [8]. However, ERBB2 is a unique member of the ERBB family, as it has a completely different extracellular structural domain from other receptors and no high-affinity ligands. ERBB2 possesses a stable conformation that mimics the ligand-activated state, with an absent domain III-V link and an exposed dimerization loop in domain II [9][10]. Structural domains I and III are in close proximity in the “open” conformation, preventing ligand binding to EGF-related peptides; this structure explains why ERBB2 lacks a ligand [9]. Notably, ERBB2 is unable to bind to any growth factor, making it impossible to induce the production of functional ERBB2/ERBB2 homodimers. Only non-physiological ERBB2 overexpression leads to the production of functional homodimers [11]. All other ERBB family members favor ERBB2 as a dimer partner and have varying signaling capacities [12][13]; in this context, homodimers have lower signaling continuity than heterodimers [14]. ERBB heterodimerization enables the incorporation of kinase-deficient ERBB3 and ligand-free ERBB2 into the signal transduction cascade. Based on this finding, this ERBB pair is believed to function as an oncogenic unit; it is the most effective in terms of contact strength, ligand-induced tyrosine phosphorylation, and downstream signaling [12][15]. Studies indicate that ERBB2 oncogenic activity from tumor ERBB2 overexpression may depend on the presence of ERBB3 [16][17].
Interestingly, ligand-induced ERBB receptor heterodimerization adheres to a rigid hierarchy. The fact that the activation of ERBB3 is hindered in the absence of ERBB2 suggests that the latter plays a part in the lateral transfer of signals between other ERBB receptors [1]. Additionally, a member of the mucin family modifies the location of ERBB2 (particularly of a phosphorylated form) in epithelial cells of the colorectum while acting as an intramembrane regulator of ERBB2 activity; this indicates that it is particularly important in the regulation of ERBB2 signaling [18]. Therefore, although none of the EGF-related peptides directly bind to ERBB2, they all cause heterodimerization and cross-phosphorylation; this in turn leads to tyrosine phosphorylation. The signal flow is accomplished via phosphorylation cascades, which begin with receptor alterations and conclude at the level of specific transcription factors.
2. ERBB2 Downstream Signaling Pathways
The characteristics of the activating ligand and the heterodimer partner are the most critical elements that decide which of the several downstream adaptor proteins will be engaged, and consequently, which pathway will be activated [14]. The C-terminal tyrosine residues of each ERBB receptor exhibit a distinctive autophosphorylation pattern, which serves as a docking site for SH2 or phosphotyrosine binding domains. Shc, Crk, Grb2, Grb7, and Gab1 are examples of adaptor proteins; Src, Chk, and phosphatidylinositol 3-kinase (PI3K; via the p85 regulatory subunit) are kinases; and SHP1 and SHP2 are protein tyrosine phosphatases. The signaling pathways triggered by the four ERBB receptors have been found to demonstrate considerable overlap. At least two important pathways are engaged in the downstream propagation of active ERBB2 signaling: the RAS/mitogen-activated protein kinase (MAPK)-dependent pathway and the PI3K-dependent pathway (Figure 2) [19]. The activation of ERBB2 signaling initiates multiple coordinated biological reactions, including mitogenesis, apoptosis, cellular motility, angiogenesis, and differentiation regulation [14].
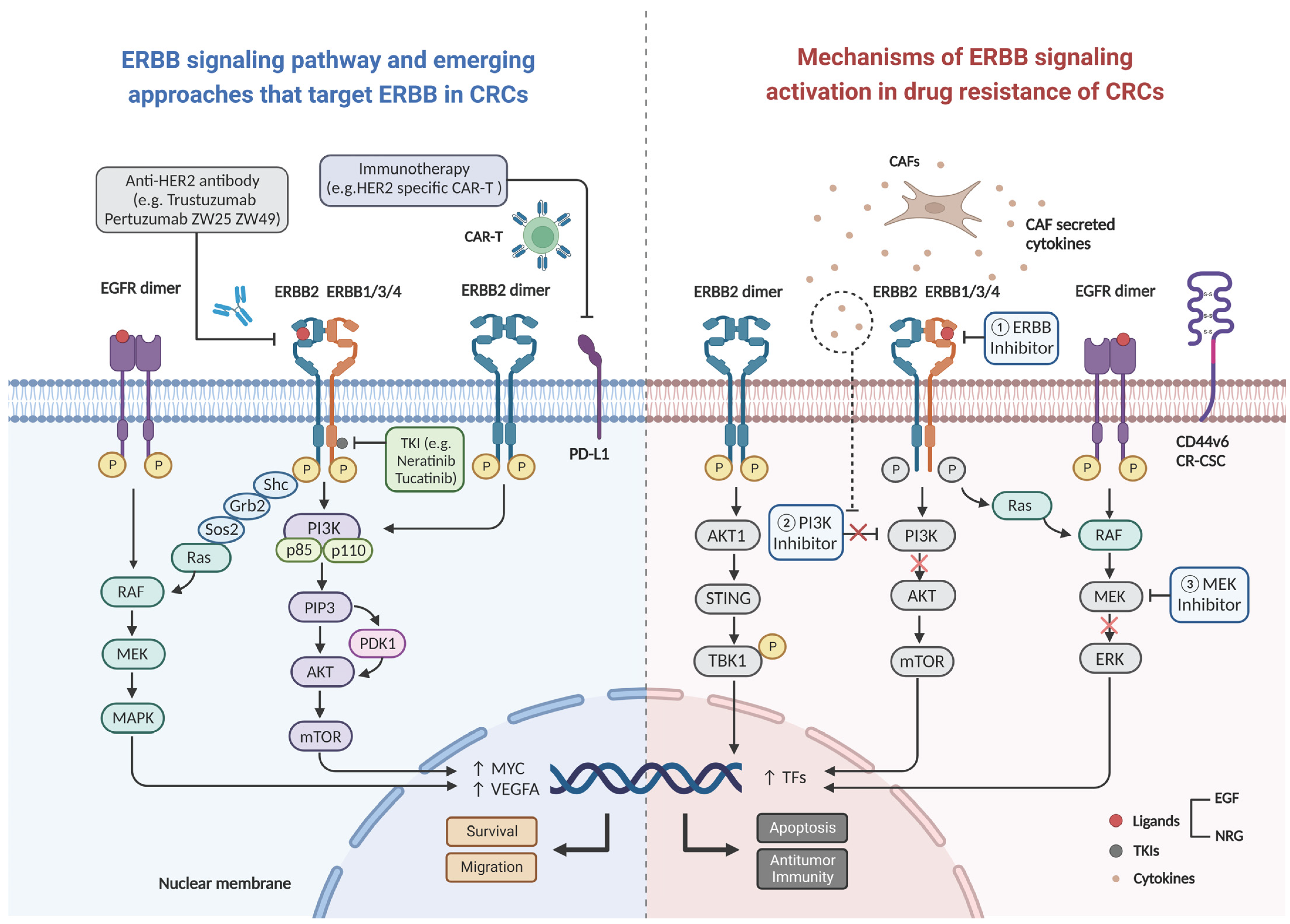
Figure 2. Schematic diagram of ligand binding activation of the ERBB2 signaling pathway leading to drug resistance. Uncontrolled formation of homo- or heterodimers, upon ERBB2 activation under physiological conditions, leads to the activation of two key downstream signaling pathways (the MAPK and PI3K-Akt pathways) and initiation of a series of cellular processes such as survival and migration. Mechanism of inhibition of the signaling pathway by various novel ERBB2 drugs (left), and the mechanisms of resistance to anti-EGFR therapy and immunotherapy due to activating of ERBB signaling pathway (right). Abbreviations: CAR-T: chimeric antigen receptor T-cell, CAFs: cancer associated fibroblasts, PD-L1: programmed cell death ligand 1, EGF: epidermal growth factor, NRG: neuregulin, CR-CSC: colorectal cancer stem cell, VEGFA: vascular endothelial growth factor A, TFs: tumor factors.
ERBB2 participates in the MAPK signaling pathway, one of the most crucial channels for cell proliferation. The three main subfamilies of MAPK include the extracellular-signal-regulated kinases (ERK MAPK, Ras/Raf1/MEK/ERK), the c-Jun N-terminal or stress-activated protein kinases (JNK or SAPK), and MAPK14 [20]. The MAP kinase enzymes are notable in that dual specificity kinases (also termed MAP/ERK kinases [MEKs] or MAP kinase kinases) need to be activated to phosphorylate both threonine and tyrosine sites [21]. Their activity is regulated by phosphorylation, which enzymatically activates MEKs. The phosphorylation state is tightly controlled by a family of proteins known as the MAP kinases (MAPKKK, MKKK, or MEKK), of which the c-Raf proto-oncogene is the most notable member [21]. Activated MAP kinases phosphorylate and activate transcription factors that are already present in the cytoplasm or nucleus; this results in the expression of certain target genes and a biological response. Responses to moderate outputs are integrated via numerous connections between several MAP kinase cascades.
The PI3K-dependent pathway is another signaling pathway associated with ERBB2. PI3Ks are divided into three classes (I–III) based on their preferred substrates and sequence homology [22]. The various isoforms within each class of PI3K and the various classes of PI3K play specific functions in cellular signal transduction. ERBB2 activates class IA PI3Ks, which are heterodimers containing a p110 catalytic subunit and a p85 regulatory subunit [23]. A lipid second messenger, namely, phosphoinositol-3,4,5-trisphosphate, binds to the pleckstrin-homology domains of numerous downstream molecules to activate them [24]; protein serine/threonine kinase AKT, commonly known as PKB, is one of its primary targets [25][26]. The mammalian targets of rapamycin-rictor kinase complex and 3-phosphoinositide-dependent kinase then recruit phosphoinositol-3,4,5-trisphosphate-bound AKT to the membrane [27], where it is subsequently phosphorylated [28]. These events lead to the full activation of AKT, which in turn phosphorylates multiple target proteins and regulates a number of cellular activities. The forkhead family of transcription factors is a significant AKT target; following phosphorylation by AKT, 14-3-3 proteins sequester them in the cytoplasm, rendering them inactive [22]. The key biological activities of the PI3K-dependent pathway include cell metabolism, cell cycle and cell survival related functions, protein synthesis, cell polarity and motility, and vesicle sorting, among others.
3. ERBB2 Gene Alterations in mCRC
The ERBB2 gene, which is found on chromosome 17q21, encodes the 185-kDa transmembrane protein ERBB2, also referred to as HER2. Expression of this protein and activation of signaling promotes a number of cellular processes including cell migration, growth, adhesion, and differentiation, that are linked to tumorigenesis [2][29]. Various solid tumors demonstrate different ERBB2 gene alterations including overexpression, amplification, and other mutations. Approximately 7% of patients with CRC have ERBB2 mutations; according to the Cancer Genome Atlas data, these are most frequently found in RAS and BRAF WT tumors [30]. In this context, ERBB2 gene amplification is the typical cause of ERBB2 protein overexpression in 4–5% of metastatic colorectal cancer (mCRC) cases [30][31][32]. Although reports are conflicting, ERBB2 overexpression has been observed more commonly in tumors with an advanced T stage and high tumor mutational burden [33]. In this context, some studies have demonstrated a substantial difference between primary tumors and metastases, indicating a decline in ERBB2 positivity with disease progression [34][35]. ERBB2 status is also related to tumor sidedness; the rectum and left colon are frequently the sites of early-stage malignancies with ERBB2 amplification, which most likely results from variables affecting germinal developmental differences [36][37][38]. However, the impact of ERBB2 amplification on the prognosis of mCRC has been debated.
Somatic activating mutations of ERBB2 have also recently been identified as contributors to the development of cancer. These mutations were first noticed in non-small cell lung cancer, followed by a number of other malignancies [39]. In all malignancies, the majority of mutations (46%) occur in the ERBB2 tyrosine kinase domain, which includes exon 20 (20%), exon 19 (11%), and exon 21 (9%) [40]. The ECD harbors 37% of ERBB2 mutations, with those of S310F/Y, Y772dupYVMA, L755P/S, V842I, and V777L/M being the most prevalent [40]. In CRC, ERBB2 mutations are most prevalent in exon 21 (23%) and the ECD (23%); the V842I variation in exon 21 is the most frequent (19%) [40]. In an in vitro study, the introduction of ERBB2 mutations at S310F, L755S, V777L, V842I, and L866M of colonic epithelial cells activated the HER2 signaling pathway and promoted non-anchored cell proliferation; this indicated that these were activating mutations [41].
Diverse ERBB2 mutations have been found in all ERBB2 gene exons including extracellular, transmembrane, or tyrosine kinase cytoplasmic domains. Downstream signaling pathways are therefore activated even in cases with normal gene copy numbers [42]. In their analysis on 111,176 tumors, Pahuja et al. found that the extracellular and kinase domains account for the majority of these changes (approximately 40% each), whereas the TMD and JMD account for 2.8% and 7.7% of the mutations, respectively [43]. In this context, G660D, R678Q, E693K, and Q709L are frequent mutations in the TMD and JMD of HER2 [43]. Pahuja et al. also evaluated the heterozygous germline HER2 TMD mutation (G660D), which was found in an Indian family; those with the mutation experienced early-onset lung cancer [43]. The majority of HER2 somatic mutations result in receptor activation, based on their capacity to boost intracellular signaling, trigger oncogenic transformation, and accelerate the growth of xenograft tumors [44][45]. However, certain HER2 mutants such as V773M have a reduced propensity for cellular activation owing to the lower quantities of phosphorylated HER2 and downstream signaling molecules that result from their synthesis [46].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14205160
References
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475.
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354.
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Rev. Rev. Biochem. 2015, 84, 739–764.
- Pawson, T.; Gish, G.D.; Nash, P. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 2001, 11, 504–511.
- Olayioye, M.A.; Graus-Porta, D.; Beerli, R.R.; Rohrer, J.; Gay, B.; Hynes, N.E. ErbB-1 and ErbB-2 acquire distinct signaling properties dependent upon their dimerization partner. Mol. Cell. Biol. 1998, 18, 5042–5051.
- Cohen, S. The epidermal growth factor (EGF). Cancer 1983, 51, 1787–1791.
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711.
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225.
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 2003, 11, 495–505.
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760.
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74.
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 1996, 16, 5276–5287.
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655.
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137.
- Pinkas-Kramarski, R.; Soussan, L.; Waterman, H.; Levkowitz, G.; Alroy, I.; Klapper, L.; Lavi, S.; Seger, R.; Ratzkin, B.J.; Sela, M.; et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996, 15, 2452–2467.
- Hsieh, A.C.; Moasser, M.M. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br. J. Cancer 2007, 97, 453–457.
- Lee-Hoeflich, S.T.; Crocker, L.; Yao, E.; Pham, T.; Munroe, X.; Hoeflich, K.P.; Sliwkowski, M.X.; Stern, H.M. A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Res. 2008, 68, 5878–5887.
- Liberelle, M.; Jonckheere, N.; Melnyk, P.; Van Seuningen, I.; Lebègue, N. EGF-Containing Membrane-Bound Mucins: A Hidden ErbB2 Targeting Pathway? J. Med. Chem. 2020, 63, 5074–5088.
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232.
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327.
- Hommes, D.W.; Peppelenbosch, M.P.; van Deventer, S.J. Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut 2003, 52, 144–151.
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657.
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619.
- DiNitto, J.P.; Cronin, T.C.; Lambright, D.G. Membrane recognition and targeting by lipid-binding domains. Sci. STKE Signal Transduct. Knowl. Environ. 2003, 2003, re16.
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C.; Toker, A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 1997, 275, 665–668.
- Klippel, A.; Kavanaugh, W.M.; Pot, D.; Williams, L.T. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol. Cell. Biol. 1997, 17, 338–344.
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101.
- Mora, A.; Komander, D.; van Aalten, D.M.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170.
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48.
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Richman, S.D.; Southward, K.; Chambers, P.; Cross, D.; Barrett, J.; Hemmings, G.; Taylor, M.; Wood, H.; Hutchins, G.; Foster, J.M.; et al. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: Analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J. Pathol. 2016, 238, 562–570.
- Loree, J.M.; Kopetz, S.; Raghav, K.P. Current companion diagnostics in advanced colorectal cancer; getting a bigger and better piece of the pie. J. Gastrointest. Oncol. 2017, 8, 199–212.
- Ingold Heppner, B.; Behrens, H.M.; Balschun, K.; Haag, J.; Krüger, S.; Becker, T.; Röcken, C. HER2/neu testing in primary colorectal carcinoma. Br. J. Cancer 2014, 111, 1977–1984.
- Lee, W.S.; Park, Y.H.; Lee, J.N.; Baek, J.H.; Lee, T.H.; Ha, S.Y. Comparison of HER2 expression between primary colorectal cancer and their corresponding metastases. Cancer Med. 2014, 3, 674–680.
- Shan, L.; Lv, Y.; Bai, B.; Huang, X.; Zhu, H. Variability in HER2 expression between primary colorectal cancer and corresponding metastases. J. Cancer Res. Clin. Oncol. 2018, 144, 2275–2281.
- Salem, M.E.; Weinberg, B.A.; Xiu, J.; El-Deiry, W.S.; Hwang, J.J.; Gatalica, Z.; Philip, P.A.; Shields, A.F.; Lenz, H.J.; Marshall, J.L. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget 2017, 8, 86356–86368.
- Loree, J.M.; Pereira, A.A.L.; Lam, M.; Willauer, A.N.; Raghav, K.; Dasari, A.; Morris, V.K.; Advani, S.; Menter, D.G.; Eng, C.; et al. Classifying Colorectal Cancer by Tumor Location Rather than Sidedness Highlights a Continuum in Mutation Profiles and Consensus Molecular Subtypes. Clin. Cancer Res. 2018, 24, 1062–1072.
- Stintzing, S.; Tejpar, S.; Gibbs, P.; Thiebach, L.; Lenz, H.J. Understanding the role of primary tumour localisation in colorectal cancer treatment and outcomes. Eur. J. Cancer 2017, 84, 69–80.
- La Salvia, A.; Lopez-Gomez, V.; Garcia-Carbonero, R. HER2-targeted therapy: An emerging strategy in advanced colorectal cancer. Expert Opin. Investig. Drugs 2019, 28, 29–38.
- Robichaux, J.P.; Elamin, Y.Y.; Vijayan, R.S.K.; Nilsson, M.B.; Hu, L.; He, J.; Zhang, F.; Pisegna, M.; Poteete, A.; Sun, H.; et al. Pan-Cancer Landscape and Analysis of ERBB2 Mutations Identifies Poziotinib as a Clinically Active Inhibitor and Enhancer of T-DM1 Activity. Cancer Cell 2019, 36, 444–457.e447.
- Kavuri, S.M.; Jain, N.; Galimi, F.; Cottino, F.; Leto, S.M.; Migliardi, G.; Searleman, A.C.; Shen, W.; Monsey, J.; Trusolino, L.; et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015, 5, 832–841.
- Zabransky, D.J.; Yankaskas, C.L.; Cochran, R.L.; Wong, H.Y.; Croessmann, S.; Chu, D.; Kavuri, S.M.; Red Brewer, M.; Rosen, D.M.; Dalton, W.B.; et al. HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc. Natl. Acad. Sci. USA 2015, 112, E6205–E6214.
- Pahuja, K.B.; Nguyen, T.T.; Jaiswal, B.S.; Prabhash, K.; Thaker, T.M.; Senger, K.; Chaudhuri, S.; Kljavin, N.M.; Antony, A.; Phalke, S.; et al. Actionable Activating Oncogenic ERBB2/HER2 Transmembrane and Juxtamembrane Domain Mutations. Cancer Cell 2018, 34, 792–806.e795.
- Greulich, H.; Kaplan, B.; Mertins, P.; Chen, T.H.; Tanaka, K.E.; Yun, C.H.; Zhang, X.; Lee, S.H.; Cho, J.; Ambrogio, L.; et al. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc. Natl. Acad. Sci. USA 2012, 109, 14476–14481.
- Wang, S.E.; Narasanna, A.; Perez-Torres, M.; Xiang, B.; Wu, F.Y.; Yang, S.; Carpenter, G.; Gazdar, A.F.; Muthuswamy, S.K.; Arteaga, C.L. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006, 10, 25–38.
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013, 3, 224–237.
This entry is offline, you can click here to edit this entry!