The study of water continues to provide surprises as evidenced by recent work on the surfaces of small droplets. Systems comprising numbers of water molecules and corresponding cations represent a topic of special interest, such as nuclear safety, functional biomolecules (DNA, RNA, and proteins) damage, proton transfer process and so on.
To date, water radical cations have been created through wet nitrogen ionized as a result of subjected to electron-impact ionization, photoionization of a water molecular beam or vapor, even including in helium nanodroplets. They have also been generated through intermolecular coulomb decay process by high-energy photos or above 70 eV electrons impactions.
For (H2O)2+•, a theoretical simulation led to both the structure resulting from proton transfer and the dimer cation structure. For larger water radical cations, these clusters are easily constructed from the (H2O)nH+ structures by substituting one of the water molecules, which is the next neighbor of the charged site for •OH radical.
Besides the ultra-fast proton transfer reaction within (H2O)n+• , and between (H2O)n+• and its neighboring water molecules to form the proton transfer product, (H2O)n+• also present ultrafast charge migration from a solute M to (H2O)n+•.
- water radical cations
- water radiolysis
- ab initio dynamics
- DFT calculations
- ultrafast chemistry
Water radical cations, (H2O)n+•, are of great research interest in both fundamental and applied sciences. Fundamental studies of water radical reactions are important to better understand the mechanisms of natural processes, such as proton transfer in aqueous solutions, the formation of hydrogen bonds and DNA damage, as well as for the discovery of new gas-phase reactions and products. In applied science, the interest in water radicals is prompted by their potential in radiobiology and as a source of primary ions for selective and sensitive chemical ionization. However, in contrast to protonated water clusters, (H2O)nH+, which are relatively easy to generate and isolate in experiments, the generation and isolation of radical water clusters, (H2O)n+•, is tremendously difficult due to their ultra-high reactivity.
1. Introduction
Water is crucial for our existence on this planet and is involved in almost all biological and chemical processes [1]. The ionization of liquid water by photons, fast electrons, X-rays, heavy ions, etc., is increasingly employed in diverse fields such as photon science, radiotherapy, nuclear reactors, radiation chemistry, nuclear waste management, and so on [2][3][4][5][6][7][8][9][10].
Following the discovery of X-rays and natural radioactive phenomena, the chemistry of water radiolysis has been extensively studied for more than a century [11][12][13]. The interaction of highly energetic photons or charged particles with water results, in general, in the ejection of a quasi-free electron from the valence shell, leaving behind a positively charged radical cation, H2O+•, which then becomes stabilized as a cluster, (H2O)n+•. Gas-phase water radical cations can also be produced in air plasma under atmospheric pressure. Experimental data suggest that the proton transfer dynamics occur on a similar timescale as electron autoionization, with the proton transfer forming a Zundel-type intermediate [HO*…H…H2O]+•, which further ionizes to form a so-far undetected type of dicationic charge-separated species with high internal energy [14].
Characterization of generated water radical cations is usually done using mass spectrometry, which can be combined with optical spectroscopy [15][16]. Two series of cluster ions ((H2O)nH+ and (H2O)n+•) in water ice have been detected simultaneously in experiments involving secondary ion mass spectrometry (SIMS), with Au+, Au3+, and C60+ as primary ions [17]. Typically, protonated (H2O)nH+ cations have been observed as the predominant products of water ionization [18][19][20]. Liu et al. recently employed a microfluidic chip combined with time-of-flight secondary ion mass spectrometry (ToF-SIMS) using keV-energy ion irradiation of Bi3+ as primary ions and only detected the protonated water and heavy water clusters [21]. The observation of radical (H2O)n+• cations is highly uncommon.
H2O+• is estimated to form within a timescale of attoseconds (10−18 s) or subfemtoseconds at the most, based on the uncertainty relationship ΔEΔt ≈ h [22]. In pure water, the hot electrons generated after the ionization of the water relax into solvent molecules and become trapped as hydrated electron (ehyd-) species, while H2O+• rapidly forms oxidizing •OH radicals via proton transfer, as shown in the following equation [23], which is fully accomplished in less than 1 ps.
|
H2O+• + H2O → •OH + H3O+ |
(1) |
In the past few decades, owing to the advent of femtosecond laser technology, several attempts using various methods have been made to identify experimentally this H2O+• cationic species [23] [24][25][26]. Nevertheless, a direct measurement of the H2O+• decay has not yet been successfully done in pure water. It is of great interest to provide insight into the electronic signature of this radical cation by using a more sophisticated time-resolved experimental or theoretical method.
Recently, Mizuse reported the infrared spectra of water cluster radical cations (H2O)n+• (n = 3–11) in the gas phase to understand the structural evolution of ionized water networks at the molecular level [15]. Further investigation has led us to have a deeper understanding of the precise structure of gaseous water radical cations (H2O)n+•. However, as has been pointed out, theoretical calculations often suffer from symmetry breaking, spin contamination and/or self-interaction errors in such open-shell doublet systems [27][28].
The reactivity of water exposed to ionizing radiation would be expected to depend on H-bond network structures around the created •OH radical, which often reacts with a substance via one-electron oxidation, addition and H abstraction. However, direct investigation of (H2O)n+• chemistry is still extremely challenging. Except for some dynamic simulations, only a few works on the oxidation of small molecules, such as O2, C2H4 or CH2O by H2O+• [29][30], and on other proton transfer and charge migration processes of H2O+•, have been reported.
The great interest in water radical cations and their solvated clusters has prompted a large number of studies which have greatly enhanced our understanding of this highly unstable transient state of water over recent years. This review focuses on recent studies aimed at uncovering the mechanism of formation, structural characterization and chemical properties of the water radical cation and its clusters.
2. Structural Properties of (H2O)n+•: Simulations and Experimental Studies
Studies of clusters have provided detailed insights into structural trends and dynamics of hydrogen-bonded water networks, as shown in Figure 1 [31][32][33][34][35][36][37][38][39][40][41][42].
Another advance in studying hydrated clusters in the gas phase is to understand chemical reactions in aqueous solutions from microscopic observations, since there are circumstances in which condensed phase studies may be unable to adequately investigate such detailed reaction mechanisms. Reducing the number of molecules in a cluster system can provide deeper insights into such a complicated system and has been used to characterize the reaction mechanism of an important chemical reaction: ionizing radiation-induced reactions of water [43][44][45][46][47][48][49][50][51][52][53].
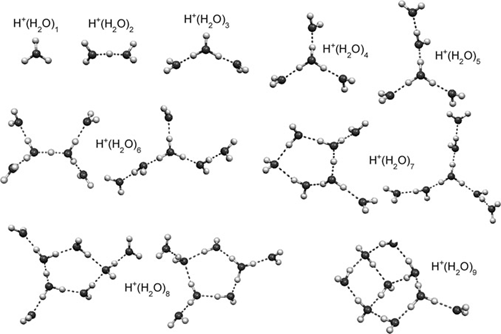
Figure 1. Experimentally characterized structures of H+(H2O)n. Figure adapted from with permission. Copyright 2011 The Royal Society of Chemistry.
Structures of (H2O)n+• would be expected to reflect the inherent instability of H2O+• and the reactivity of cationized water networks. Attaining such knowledge is quite important for understanding ionizing-radiation-induced chemistry in water. However, although (H2O)n+• has been produced in trace amounts in several experimental studies, structural information on these species is still limited. This section focuses on this problem.
3. Chemical Properties of (H2O)n+•: Simulations and Experimental Studies
3.1. Proton Transfer to Form Hydroxyl Radicals
Ionization of water clusters (liquid, molecular beam or vapor) has been investigated using several techniques, and the reaction was indicated according to the results to follow the equations
|
(H2O)n + Ip → [(H2O)n+•]ver + e− (Ionization) |
(6) |
|
[(H2O)n+•]ver → (H3O+)(•OH)(H2O)n−2 (Complex Formation) |
(7) |
where Ip and ver denote the ionization potential and vertical ionized state from the parent neutral cluster, respectively. Based on a previous study in which the ionizations of water clusters (H2O)n (n = 2–6) were investigated using the direct ab initio molecular dynamics (AIMD) method at the Hartree–Fock (HF) level [117], Tachikawa et al. performed the direct AIMD calculation on a more accurate potential energy surface (MP2/6-311++G(d,p) level) to estimate the rate of proton transfer along the hydrogen bond in the water cluster cation [118]. The rate of the first proton transfer was found to be strongly dependent on the cluster size: average time scales of proton transfer for n = 2, 3 and 4 were 28, 15 and 10 fs, respectively (from the MP2/6-311++G (d,p) level calculation), suggesting the proton transfer reactions to be very rapid processes in the three clusters. The clusters with n = 3 and 4 also each showed a second proton transfer (with average time scales of 120 fs and 40 fs, respectively, after the ionization), as shown in Figures 2 and 3 for the case of the water tetramer (H2O)4+•.
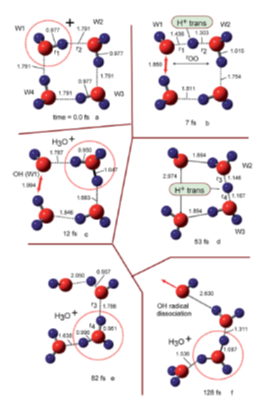
Figure 2. Snapshots of water tetramer cation (H2O)4+• after vertical ionization from optimized structure. The values are bond distance in Å. Figure adapted from with permission. Copyright 2015 The Royal Society of Chemistry.
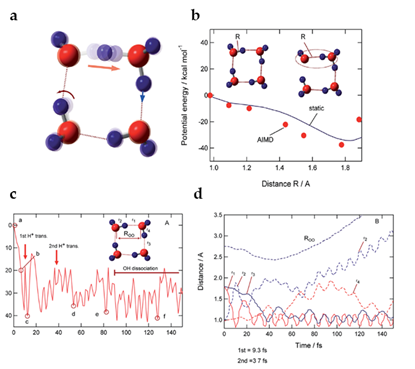
Figure 3. (a) Superposition of snapshots for the first proton transfer process. (b) Potential energy curve for the proton transfer process as derived from a static ab initio calculation (MP2/6-311++G (d, p) level). The values obtained from the direct ab initio molecular dynamics (AIMD) calculation are given as filled circles. (c) Potential energy and (d) geometrical parameters of the water tetramer cation following the vertical ionization each as a function of time. Figure adapted from with permission. Copyright 2015 The Royal Society of Chemistry.
As illustrated above, on the basis of the presented calculations, a reaction model of ionizations of the water clusters is proposed. Upon ionization of water, a hole would become localized on one of the water molecules of (H2O)n, and a proton would rapidly transfer—within a time scale of about 10 fs—from H2O+• (W1+•) to H2O (W2) along the hydrogen bond. Next, the second proton transfer would occur from W2 (H+) to W3 (H2O) within a time scale of 50–100 fs. The second proton transfer would result in the separation of the •OH radical from H3O+ to form H3O+-H2O-•OH, as discussed in detail above in Section 3. Upon completion of this separation, any attraction between H3O+ and the •OH radical would be masked by the intervening H2O, immediately resulting in dissociation of the •OH radical from the system.
3.2. Initial Ultrarapid Charge Migration in the Chemistry of H2O+•
Generally, the •OH radical dissociated from H2O+• is considered to be the main reactive species inducing one electron oxidation, •OH adduct formation and H atom abstraction reactions. In theory, H2O+• should be considered as an ultra-oxidizing species. In the gas phase, oxidation of small molecules such as O2 has been reported, as discussed above. In the condensed phase, a prerequisite for the direct oxidation process to be competitive is that the target molecule should be in close proximity to H2O+•; thus, the oxidation process induced by H2O+• can be mimicked in highly concentrated solutions, where the nearest neighbors of H2O+• may be molecules other than water [54][55]. By effectively solving the bottleneck, it was not possible to exclude the possibility of an •OH radical reaction in the oxidation process by using nanosecond or microsecond electron pulses as well as the problems of the absence of information on the yield of a direct effect of radiation on the solute, picosecond pulse radiolysis clearly showed that H2O+• can also be considered as an ultra-oxidizing species in solution and presents a reactivity different from that of the •OH radical. Furthermore, several attempts have been made to experimentally investigate H2O+• in different ways based on femtosecond laser technology.
One of the femtosecond laser-driven accelerators established at ELYSE (a facility named after Lysis (Greek for degradation) by Electrons, which achieves a high energy (7-8 Mev) electron beam with a pulse width of 7 ps) is shown in Figure 4. This accelerator can achieve a high-energy (7–8 MeV) electron beam with a pulse width of 7 ps [56]. Its detection system is based on transient absorption spectroscopy with probe light, with wavelengths ranging from 380 nm to 1500 nm [57]. Therefore, the unique time-resolved technique based on a high-energy electron pulse enabled various investigators to explore ultra-rapid chemical reactions of H2O+• through the scavenging method in a variety of highly concentrated aqueous solutions, i.e., by observing the formation of secondary radicals such as NO3•, SO4•−, X2•− (X = Cl, Br) or H2PO4•. By further using diffusion-kinetic simulations of the spur reaction induced by the incident electrons, the first semi-quantitative estimation of the H2O+• radicals scavenging fractions for a wide range of solutes could be established.
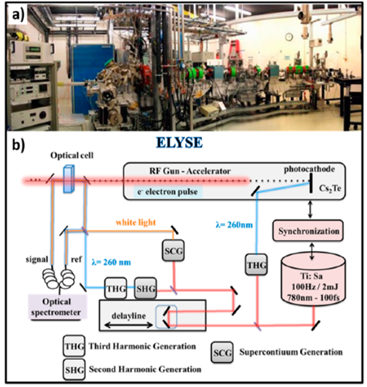
Figure 4. (a) Photograph of the pulse radiolysis facility (the only one in Europe) based on the ELYSE (a facility named after Lysis (Greek for degradation) by Electrons, which achieves a high energy (7-8 Mev) electron beam with a pulse width of 7 ps)picosecond pulsed electron accelerator from the Physical Chemistry Laboratory in Orsay, France. (b) Schematic description of the synchronization of the electron beam for ionization with a laser beam to probe the species created by the electron pulse. Figure adapted from [58] with permission. Copyright 2018 Multidisciplinary Digital Publishing Institute.
H2O+• has been suggested to be a stronger oxidative radical than other oxidants in aqueous solutions based on an estimation of its redox potential value: the standard redox potential of H2O+•/H2O couple has been recently estimated to be higher than 3 V vs. normal hydrogen electrode (NHE) [58]. A 7 ps pulse radiolysis set-up was used to demonstrate the oxidation reaction of H2O+• in various solutions. As the lifetime of H2O+• is too short for it to be observed directly, the product of the oxidation reaction was observed just after the electron pulse. The oxidation of M by H2O+• and by direct effect gives M+•, which can be observed. Other oxidation reactions, such as oxidation by the •OH radical, do not occur so quickly. Therefore, the yield of M+• measured precisely at 7 ps could provide information about its oxidation by H2O+• (as shown in Figure 5).
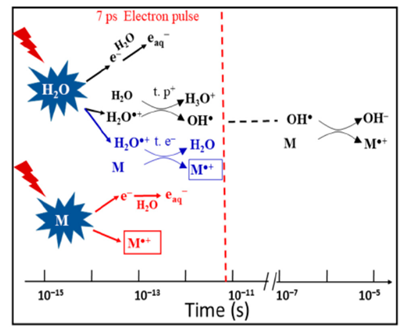
Figure 5. Schematic description of the reactions occurring in solutions containing a solute M at high concentration. Figure adapted from [58] with permission. Copyright 2018 Multidisciplinary Digital Publishing Institute.
Several studies in highly concentrated solutions were performed with the purpose of scavenging the radical cation of water, i.e., H2O+• [59][60][61][62][63]. Highly concentrated sulfuric acid solutions were shown to be appropriate probe systems to elucidate the different mechanisms [64][65][66][67][68][69] and showed H2O+• acting as a strong oxidant towards weak electron donors such as HSO4− or H2PO4− [70]. The reactivities of H2O+• and D2O+• were probed in hydrogenated and deuterated sulfuric acid solutions of various concentrations, combined with theoretical simulations by Wanget al. [71]
As illustrated in Figures 6 and 7, electron transfer was indicated, based on the kinetics observed by Wanget al. [136], to be favored over proton transfer in competition reactions of the radical cation of water in deuterated solutions. These latest studies thus indicated the radical cation of water to be engaged not only in proton transfer reactions but also in redox reactions when H2O+• (D2O+•) is formed in the vicinity of molecules different from water—with charge migration propelled by the excess energy present in the electron cloud just after ionization and not by nuclear motions as in standard chemical theories of electron transfer [71][72].
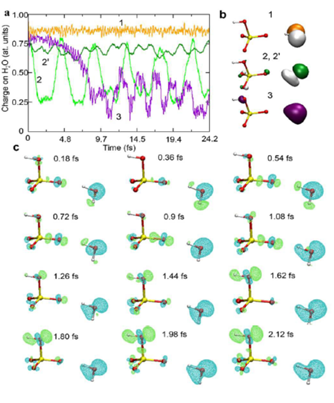
Figure 6. Electronic dynamics simulations. (a) Evolution of the charge of the water molecule hydrogen bonded to HSO4- as a function of time after ionization took place, defined to be t = 0. Each plot (1, 2, 2′ and 3) corresponds to a different electronic dynamics generated by depopulating different valence MOs, the representations of which are shown in panel (b) for isosurfaces of ± 0.08 bohr−3/2. The two green curves (2 and 2′) correspond to ionization from the same MO but with different values of the hydrogen bond length between H2O and HSO4−, 1.8 and 2.4 Å, respectively, and (c) isosurfaces of the difference charge density with respect to the initial time for dynamics 2 and for different times spanning the first half period of charge migration. Blue and green isosurfaces indicate accumulation and depletion of electron density, respectively. Figure adapted from [71] with permission. Copyright 2017 The Royal Society of Chemistry.
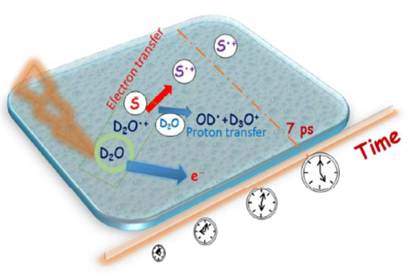
Figure 7. Schematic of the probing of the competition reactions of the radical cation D2O+•. A time resolution of 7 ps was used in the setup for probing the sulfate radical in deuterated sulfuric acid solutions. Figure adapted from [71] with permission. Copyright 2017 The Royal Society of Chemistry.
This entry is adapted from the peer-reviewed paper 10.3390/molecules25153490
References
- Bellissent-Funel, M.C.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; van der Spoel, D.; Xu, Y.; Garcia, A.E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697, doi:10.1021/acs.chemrev.5b00664.
- Mizuse, K.; Mikami, N.; Fujii, A. Infrared Spectra and Hydrogen-Bonded Network Structures of Large Protonated Water Clusters H+(H2O)n (n = 20–200). Angew. Chem. Int. Ed. 2010, 49, 10119–10122, doi:10.1002/anie.201003662.
- Lehnert, S. Biomolecular Action of Ionizing Radiation; Taylor & Francis Group: Oxfordshire, UK, 2008.
- Narcisi, R.S.; Bailey, A.D. Mass Spectrometric Measurements of Positive Ions at Altitudes from 64-112 kilometers. J. Geophys. Rsc. 1965, 70, 3687–3700.
- Arnold, F.; Krankowsky, D.; Marien, K.H. First Mass Spectrometric Measurements of Positive Ions in the Stratosphere. Nature 1977, 267, 30–31.
- Smith, D.; Spanel, P. Ions in the terrestrial atmosphere and in interstellar clouds. Mass Spectrom. Rev. 1995, 14, 255–278.
- Draganic, I.G.; Draganic, Z.D. The Radiation Chemistry of Water; Academic Press: New York, 1971.
- Alizadeh, E.; Sanche, L. Precursors of Solvated Electrons in Radiobiological Physics and Chemistry. Chem. Rev. 2012, 112, 5578–5602, doi:10.1021/cr300063r.
- Ceppatelli, M.; Bini, R.; Schettino, V. High-pressure photodissociation of water as a tool for hydrogen synthesis and fundamental chemistry. PNAS USA 2009, 106, 11454–11459, doi:10.1073/pnas.0901836106.
- Shinohara, H.; Nishi, N.; Washida, N. Photoionization of water clusters at 11.83 eV: Observation of unprotonated cluster ions (H2O)+n (2 ≤ n ≤ 10). J. Chem. Phys. 1986, 84, 5561–5567, doi:10.1063/1.449914.
- Hatano, Y.; Mozumder, A. Charged Particle and Photo Interactions with Matter; CRC Press: Boca Raton, FL, USA, 2003.
- Garrett, B.C.; Dixon, D.A.; Camaioni, D.M.; Chipman, D.M.; Johnson, M.A.; Jonah, C.D.; Kimmel, G.A.; Miller, J.H.; Rescigno, T.N.; Rossky, P.J., et al. Role of Water in Electron-Initiated Processes and Radical Chemistry Issues and Scientific Advances. Chem. Rev. 2005, 105, 355–390.
- Buxton, G.V. Radiation Chemistry-From Basics to Applications in Material and Life Sciences; EDP Sciences: Les Ulis, France, 2008.
- Thurmer, H.; Oncak, M.; Ottosson, N.; Seidel, R.; Hergenhahn, U.; Bradforth, S.E.; Slavicek, P.; Winter, B. On the Nature and Origin of Dicationic, Charge-separated Species Formed in Liquid Water on X-ray Irradiation. Nat. Chem. 2013, 5, 590–596, doi:doi.org/10.1038/nchem.1680.
- Mizuse, K.; Kuo, J.-L.; Fujii, A. Structural trends of ionized water networks: Infrared spectroscopy of watercluster radical cations (H2O)n+ (n = 3–11). Chem. Sci. 2011, 2, 868–876, doi:10.1039/c0sc00604a.
- Jiang, J.C.; Wang, Y.S.; Chang, H.C.; Lin, S.H.; Lee, Y.T.; Niedner-Schatteburg, G.; Chang, H.C. Infrared Spectra of H+(H2O)5-8 Clusters Evidence for Symmetric Proton Hydration. J. Am. Chem. Soc. 2000, 122, 1398–1410.
- Conlan, X.A.; Fletcher, J.S.; Lockyer, N.P.; Vickerman, J.C. A Comparative Study of Secondary of Secondary Ion Emission from Water Ice under Ion Bombardment by Au+, Au3+, and C60+. J. Phys. Chem. C 2010, 114, 5468–5479.
- Good, A.; Durden, D.A.; Kebarle, P. Ion-molecule Reactions in Pure Nitrogen and Nitrogen Containing Traces of Water at Total Pressures 0.5-4 torr. Kinetics of Clustering Reaction Forming H+(H2O)n. J. Chem. Phys. 1970, 52, 212–221.
- Good, A.; Durden, D.A.; Kebarle, P. Mechanism and Rate Constants of Ion-molecule Reactions Leading to Formation of H+(H2O)n in Moist Oxygen and Air. J. Chem. Phys. 1970, 52, 222–229.
- Shahin, M.M. Mass-Spectrometric Studies of Corona Discharges in Air at Atmospheric Pressures. J. Chem. Phys. 1966, 45, 2600–2605, doi:10.1063/1.1727980.
- Liu, Y.-Y.; Ying, Y.-L.; Hua, X.; Long, Y.-T. In-situ discrimination of the water cluster size distribution in aqueous solution by ToF-SIMS. Sci. China Chem. 2017, 61, 159–163, doi:10.1007/s11426-017-9180-1.
- Mozumder, A. Events in Radiation Chemistry An Introduction. Int. J. Radiat. Appl. Instrum. Part. C 1989, 34, 1–3.
- Gauduel, Y.; Pommeret, S.; Migus, A.; Antonetti, A. Some Evidence of Ultrafast H2O+.-Water Molecule Reaction in Femosecond Photoionizaiton of Pure Liquid Water: Influence on Geminate Pair Recombination Dynamics. Chem. Phys. 1990, 149, 1–10.
- Long, F.H.; Lu, H.; Eisenthal, K.B. Femtosecond studies of the presolvated electron: An excited state of the solvated electron? Phys. Rev. Lett. 1990, 64, 1469–1472, doi:10.1103/PhysRevLett.64.1469.
- Marsalek, O.; Elles, C.G.; Pieniazek, P.A.; Pluharova, E.; VandeVondele, J.; Bradforth, S.E.; Jungwirth, P. Chasing charge localization and chemical reactivity following photoionization in liquid water. J. Chem. Phys. 2011, 135, 224510, doi:10.1063/1.3664746.
- Li, J.; Nie, Z.; Zheng, Y.Y.; Dong, S.; Loh, Z.-H. Elementary Electron and Ion Dynamics in Ionized Liquid Water. J. Chem. Phys. Lett. 2013, 4, 3698–3703, doi:10.1021/jz401987f.
- Kamarchik, E.; Kostko, O.; Bowman, J.M.; Ahmed, M.; Krylov, A.I. Spectroscopic signatures of proton transfer dynamics in the water dimer cation. J. Chem. Phys. 2010, 132, 194311, doi:10.1063/1.3432198.
- Sodupe, M.; Bertran, J.; Rodriguez-Santiago, L.; Baerends, E.J. Ground State of the (H2O)n+. Radical Cation DFT versus Post-Hartree-Fock Methods. J. Phys. Chem. A 103, 166-170.
- Karpas, Z.; Huntress Jr, W.T. Reactions of OH+ and H2O+. ions with some diatomic and simple polyatomic molecules. Chem. Phys. Lett. 1978, 59, 87–89.
- Dotan, I.; Lindinger, W.; Rowe, B.; Fahey, D.W.; Fehsenfeld, F.C.; Albritton, D.L. Rate Constants for the Reaction of H2O+. with NO2, O2, NO, C2H4, CO, CH4, and H2 measured at relative kinetic energies 0.04-2 eV. Chem. Phys. Lett. 1980, 72, 67–70.
- Sato, K.; Tomoda, S.; Kimura, K.; Iwata, S. Electronic and Molecular Structure of the Water Dimer Cation. A Theoretical Study. Chem. Phys. Lett. 1983, 95, 579–583.
- De Visser, S.P.; De Koning, L.J.; Nibbeering, N.N.M. Reactivity and Thermochemical Properties of the Water Dimer Radical Cation in the Gas Phase. J. Phys. Chem. 1995, 99, 15444–15447.
- Yang, X.; Castleman, A.W. Large Protonated Water Clusters H+(H2O)n ( 1. Ltoreq. N > 60): The Production and Reactivity of Clathrate-like Structures Under Thermal Conditions. J. Am. Chem. Soc. 1989, 111, 6845–6846.
- Wei, S.; Shi, Z.; Castleman, J.A.W. Mixed Cluster Ions as a Structure Probe: Experimental Evidence for Clathrate Structure of (H2O)20H+ and (H2O)21H+. J. Chem. Phys. 1991, 94, 3268–3270.
- Dalleska, N.F.; Honma, K.; Armentrout, P.B. Stepwise Solvation Enthalpies of Protonated Water Clusters: Collision-Induced Dissociation as an Alternative to Equilibrium Studies. J. Am. Chem. Soc. 1993, 115, 12125–12131.
- Hansen, K.; Andersson, P.U.; Uggerud, E. Activation Energies for Evaporation From Protonated and Deprotonated Water Clusters From Mass Spectra. J. Chem. Phys. 2009, 131, 124303.
- Hulthe, G.; Stenhagen, G.; Wennerstrom, O.; Ottosson, C.-H. Water Clusters Studied by Electrospray Mass Spectrometry. J. Chromatogr. A 1997, 777, 155–156.
- Haberland, H.; Ludewigt, C.; Schindler, H.G.; Worsnop, D.R. Experimental observation of the negatively charged water dimer and other small (H2O)−n clusters. J. Chem. Phys. 1984, 81, 3742–3744, doi:10.1063/1.448127.
- Lin, C.K.; Wu, C.C.; Wang, Y.S.; Lee, Y.T.; Chang, H.C.; Kuo, O.J.; Klein, M.L. Vibrational predissociation spectra and hydrogen-bond topologies of H+(H2O)9–11. Phys. Chem. Chem. Phys. 2005, 7, 938–944, doi:10.1039/b412281j.
- Lisy, J.M. Infrared studies of ionic clusters: The influence of Yuan T. Lee. J. Chem. Phys. 2006, 125, 132302, doi:10.1063/1.2338317.
- Okumura, M.; Yeh, L.I.; Myers, J.D.; Lee, Y.T. Infrared Spectra of the Solvated Hydronium Ion: Vibrational Predissociation Spectroscopy of Mass-Selected H3O+•(H2O)n•(H2)m. J. Phys. Chem. 1990, 94, 3146–3427.
- Mizuse, K.; Fujii, A. Infrared Photodissociation Spectroscopy of H+(H2O)6•Mm (M = Ne, Ar, Kr, Xe, H2, N2, and CH4): Messenger-Dependent Balance Between H3O+ and H5O2+ Core Isomers. Phys. Chem. Chem. Phys. 2011, 13, 7129–7135.
- Petrenko, V.F.; Khusnatdinov, N.N. On the nature of photo charge carriers in ice. J. Chem. Phys. 1994, 100, 9096–9105, doi:10.1063/1.466663.
- Narten, A.H.; Levy, H.A. Liquid Water: Molecular Correlation Functions from X-Ray Diffraction. J. Chem. Phys. 1971, 55, 2263–2269, doi:10.1063/1.1676403.
- Castleman, A.W.; Bowen, K.H. Clusters: Structure, Energetics, and Dynamics of Intermediate States of Matter. J. Phys. Chem. 1996, 100, 12911–12944.
- Niedner-Schatteburg, G.; Bondybey, V.E. FT-ICR Studies of Solvation Effects in Ionic Water Cluster Reactions. Chem. Rev. 2000, 100, 4059–4086, doi:10.1021/cr990065o.
- Bondybey, V.E.; Beyer, M.K. How many molecules make a solution? Inter. Rev. Phys. Chem. 2010, 21, 277–306, doi:10.1080/01442350210132741.
- Mohammed, O.F.; Pines, D.; Dreyer, J.; Pines, E.; Nibbering, E.T. Sequential proton transfer through water bridges in acid-base reactions. Science 2005, 310, 83–86, doi:10.1126/science.1117756.
- Headrick, J.M.; Diken, E.G.; Walters, R.S.; Hammer, N.I.; Christie, R.A.; Cui, J.; Myshakin, E.M.; Duncan, M.A.; Johnson, M.A.; Jordan, K.D. Spectral signatures of hydrated proton vibrations in water clusters. Science 2005, 308, 1765–1769, doi:10.1126/science.1113094.
- Eigen, M.; de Maeyer, L. Self-dissociation and protonic charge transport in water and. Proc. Math. Phys. Eng. Sci. 1958, 247, 505–533, doi:10.1098/rspa.1958.0208.
- Eigen, M. Proton Transfer, Acid-Base Catalysis, and Enzymatic Hydrolysis. Angew. Chem. Int. Ed. 1964, 3, 1–19.
- Marx, D.; Tuckerman, M.E.; Hutter, J.; Parrinello, M. The Nature of the Hydrated Excess Proton. Nature 1999, 397, 601–604.
- de Grotthuss, C.J.T. Memoir on the Decomposition of Water and of the Bodies That It Holds in Solution by Means of Galvanic Electricity. Biochim. Biophys. Acta 2006, 1957, 871–875.
- Khorana, S.; Hamill, W.H. Electronic Processes in the Pulse Radiolysis of Aqueous Solutions of Halide Ions. J. Phys. Chem. 1971, 75, 3081–3088.
- Kim, K.J.; Hamill, W.H. Direct and Indirect Effects in Pulse Irradiated Concentrated Aqueous Solutions of Chloride and Sulfate Ions. J. Phys. Chem. 1976, 80, 2320–2325.
- Belloni, J.; Monard, H.; Gobert, F.; Larbre, J.P.; Demarque, A.; De Waele, V.; Lampre, I.; Marignier, J.L.; Mostafavi, M.; Bourdon, J.C., et al. ELYSE—A picosecond electron accelerator for pulse radiolysis research. Nucl. Instrum. Methods Phys. Res. Sec. A 2005, 539, 527–539, doi:10.1016/j.nima.2004.11.006.
- Schmidhammer, U.; Jeunesse, P.; Stresing, G.; Mostafavi, M. A Broadband Ultrafast Transient Absorption Spectrometer Covering the Range from Near-Infrared (NIR) Down to Green. Appl. Spectrosc. 2014, 68, 1137–1147.
- Ma, J.; Wang, F.; Mostafavi, M. Ultrafast Chemistry of Water Radical Cation, H(2)O(*+), in Aqueous Solutions. Molecules 2018, 232, 244, doi:10.3390/molecules23020244.
- Balcerzyk, A.; LaVerne, J.; Mostafavi, M. Direct and indirect radiolytic effects in highly concentrated aqueous solutions of bromide. J. Phys. Chem. A 2011, 115, 4326–4333, doi:10.1021/jp2012528.
- EI Omar, A.K.; Schmidhammer, U.; Rousseau, B.; Laverne, J.; Mostafavi, M. Competition Reaction of H2O+. Radical in Concentrated Cl- Aqueous Solutions: Picosecond Pulse Radiolysis Study. J. Phys. Chem. A 2012, 116, 11509–11518.
- Balcerzyk, A.; Schmidhammer, U.; El Omar, A.K.; Jeunesse, P.; Larbre, J.P.; Mostafavi, M. Picosecond Pulse Radiolysis of Direct and Indirect Radiolytic Effects in Highly Concentrated Halide Aqueous Solutions. J. Phys. Chem. A 2011, 115, 9151–9159.
- El Omar, A.K.; Schmidhammer, U.; Balcerzyk, A.; La Verne, J. Spur Reactions Observed by Picosecond Pulse Radiolysis in Highly Concentrated Bromide Aqueous Solutions. J. Phys. Chem. A 2013, 117, 2287–2293.
- Ghalei, M.; Ma, J.; Schmidhammer, U.; Vandenborre, J.; Fattahi, M.; Mostafavi, M. Picosecond Pulse Radiolysis of Highly Concentrated Carbonate Solutions. J. Phys. Chem. B 2016, 120, 2434–2439, doi:10.1021/acs.jpcb.5b12405.
- Ma, J.; Schmidhammer, U.; Mostafavi, M. Picosecond Pulse Radiolysis of Highly Concentrated Sulfuric Acid Solutions: Evidence for the Oxidation Reactivity of Radical Cation H2O+. J. Phys. Chem. A 2014, 118, 4030–4037.
- Choe, Y.K.; Tsuchida, E.; Ikeshoji, T. First-principles molecular dynamics study on aqueous sulfuric acid solutions. J. Chem. Phys. 2007, 126, 154510, doi:10.1063/1.2718526.
- Kameda, Y.; Hosoya, K.; Sakamoto, S.; Suzuki, H.; Usuki, T.; Uemura, O. Hydrogen-Bonded Structure in Aqueous Sulfuric Acid Solutions. J. Mol. Liq. 1995, 65, 305–308.
- Ma, J.; Schmidhammer, U.; Pernot, P.; Mostafavi, M. Reactivity of the Strongest Oxidizing Species in Aqueous Solutions: The Short-Lived Radical Cation H2O+. J. Phys. Chem. Lett. 2014, 5, 258–261.
- Tang, Y.; Thorn, R.P.; Mauldin, R.L.; Wine, P.H. Kinetics and Spectroscopy of SO4- Radical in Aqueous Solution. J. Photochem. Photobiol. A 1988, 44, 243–258.
- Hayon, E.; Treinin, A.; Wilf, J. Electronic Spectra, Photochemistry, and Autoxidation Mechanism of the Sulfite-Bisulfite-Pyrosulfite Systems. SO2-, SO3-, SO4-, and SO5- Radicals. J. Am. Chem. Soc. 1972, 94, 47–57.
- Ma, J.; Schmidhammer, U.; Mostafavi, M. icosecond Pulse Radiolysis of Highly Concentrated Phosphoric Acid Solutions: Mechanism of Phosphate Radical Formation. J. Phys. Chem. B 2015, 119, 7180–7185.
- Wang, F.; Schmidhammer, U.; Lande, A.; Mostafavi, M. Ultra-Fast Charge Migration Competes with Proton Transfer in the Early Chemistry of H2O+. PCCP 2017, 19, 2894–2899, doi:10.1039/C6CP07013B.
- Lépine, F.; Ivanov, M.Y.; Vrakking, M.J.J. Attosecond molecular dynamics: Fact or fiction? Nat. Photonics 2014, 8, 195–204, doi:10.1038/nphoton.2014.25.