A tumor suppressor gene, or anti-oncogene, is a gene that regulates a cell during cell division and replication. If the cell grows uncontrollably, it will result in cancer. When a tumor suppressor gene is mutated, it results in a loss or reduction in its function; in combination with other genetic mutations this could allow the cell to grow abnormally. The loss of function for these genes may be even more significant in the development of human cancers, compared to the activation of oncogenes. Tumor suppressor genes can be grouped into the following categories caretaker genes, gatekeeper genes, and more recently landscaper genes. Caretaker genes ensure stability of the genome via DNA repair and subsequently when mutated allow mutations to accumulate. Meanwhile gatekeeper genes directly regulate cell growth by either inhibiting cell cycle progression or inducing apoptosis. Lastly landscaper genes regulate growth by contributing to the surrounding environment, when mutated can cause an environment that promotes unregulated proliferation. The classification schemes are evolving as medical advances are being made from fields including molecular biology, genetics, and epigenetics.
- cell division
- epigenetics
- cell cycle progression
1. Two-hit Hypothesis
Unlike oncogenes, tumor suppressor genes generally follow the two-hit hypothesis, which states both alleles that code for a particular protein must be affected before an effect is manifested[1]. If only one allele for the gene is damaged, the other can still produce enough of the correct protein to retain the appropriate function. In other words, mutant tumor suppressor alleles are usually recessive, whereas mutant oncogene alleles are typically dominant.
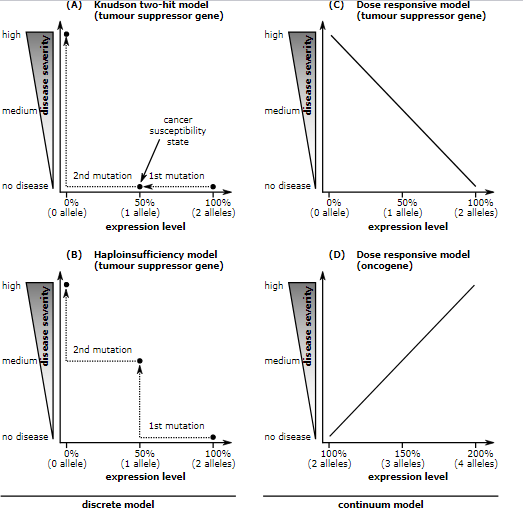
The two-hit hypothesis was first proposed by A.G. Knudson for cases of retinoblastoma.[1] He observed that 40% of U.S cases were caused by a mutation in the germ-line. However, affected parents could have children without the disease; but the unaffected children became parents of children with retinoblastoma.[2] This indicates that one could inherit a mutated germ-line but not display the disease. Knudson observed that the age of onset of retinoblastoma followed 2nd order kinetics, implying that two independent genetic events were necessary. He recognized that this was consistent with a recessive mutation involving a single gene, but requiring bi-allelic mutation. Hereditary cases involve an inherited mutation and a single mutation in the normal allele.[2] Non-hereditary retinoblastoma involves two mutations, one on each allele.[2] Knudson also noted that hereditary cases often developed bilateral tumors and would develop them earlier in life, compared to non-hereditary cases where individuals were only affected by a single tumor.[2]
There are exceptions to the two-hit rule for tumor suppressors, such as certain mutations in the p53 gene product. p53 mutations can function as a dominant negative, meaning that a mutated p53 protein can prevent the function of the natural protein produced from the non-mutated allele.[3] Other tumor-suppressor genes that do not follow the two-hit rule are those that exhibit haploinsufficiency, including PTCH in medulloblastoma and NF1 in neurofibroma. Another example is p27, a cell-cycle inhibitor, that when one allele is mutated causes increased carcinogen susceptibility.[4]
2. Functions
Tumor-suppressor genes, or more precisely, the proteins for which they encode, can have a repressive effect on the regulation of the cell cycle, promote apoptosis, or sometimes both. Tumor-suppressor proteins function in various ways, which include the following:[5]
- Repression of genes that are essential for continuing the progression of the cell cycle. If these genes are not expressed, the cell cycle does not continue, effectively inhibiting cell division.
- Coupling the cell cycle to DNA damage. As long as there is damaged DNA in the cell, it should not divide. If the damage can be repaired, the cell cycle can continue.
- Apoptosis. If damage cannot be repaired, the cell should initiate apoptosis (programmed cell death) to remove the threat it poses to the organism as a whole.
- Cell adhesion. Some proteins involved in cell adhesion prevent tumor cells from dispersing, block loss of contact inhibition, and inhibit metastasis. These proteins are known as metastasis suppressors.[6][7]
- DNA repair. Caretaker genes encode proteins that function in repairing mutations in the genome, preventing cells from replicating with mutations. Furthermore, increased mutation rate from decreased DNA repair leads to increased inactivation of other tumor suppressors and activation of oncogenes.[8]
3. Examples
There are many different tumor suppressor genes, including
- Retinoblastoma protein (pRb). pRb was the first tumor-suppressor protein discovered in human retinoblastoma; however, recent evidence has also implicated pRb as a tumor-survival factor. RB1 gene is a gatekeeper gene that blocks cell proliferation, regulates cell division and cell death[2]. Specifically pRb prevents the cell cycle progression from G1 phase into the S phase by binding to E2F and repressing the necessary gene transcription.[9] This prevents the cell from replicating its DNA if there is damage.
- p53. TP53, a caretaker gene, encodes the protein p53, which is nick named "the guardian of the genome". p53 has many different functions in the cell including DNA repair, inducing apoptosis, transcription, and regulating the cell cycle.[10] Mutated p53 is involved in many human cancers, of the 6.5 million cancer diagnoses each year about 37% are connected to p53 mutations[10]. This makes it a popular target for new cancer therapies. Homozygous loss of p53 is found in 65% of colon cancers, 30–50% of breast cancers, and 50% of lung cancers. Mutated p53 is also involved in the pathophysiology of leukemias, lymphomas, sarcomas, and neurogenic tumors. Abnormalities of the p53 gene can be inherited in Li-Fraumeni syndrome (LFS), which increases the risk of developing various types of cancers.
- BCL2. BCL2 is a family of proteins that are involved in either inducing or inhibiting apoptosis.[11] The main function is involved in maintaining the composition of the mitochondria membrane, and preventing cytochrome c release into the cytosol[11]. When cytochrome c is released from the mitochondria it starts a signaling cascade to begin apoptosis.[12]
- SWI/SNF. SWI/SNF is a chromatin remodelling complex, which is lost in about 20% of tumors.[13] The complex consists of 10-15 subunits encoded by 20 different genes[13]. Mutations in the individual complexes can lead to misfolding, which compromises the ability of the complex to work together as a whole. SWI/SNF has the ability move nucleosomes, which condenses DNA, allowing for transcription or block transcription from occurring for certain genes[13]. Mutating this ability could cause genes to be turned on or off at the wrong times.
As the cost of DNA sequencing continues to diminish, more cancers can be sequenced. This allows for the discovery of novel tumor suppressors and can give insight on how to treat and cure different cancers in the future. Other examples of tumour suppressors include pVHL, APC, CD95, ST5, YPEL3, ST7, and ST14, p16, BRCA2.[14].
The content is sourced from: https://handwiki.org/wiki/Biology:Tumor_suppressor
References
- Knudson AG (1971). "Mutation and Cancer: Statistical Study of Retinoblastoma". Proc Natl Acad Sci USA 68 (4): 820–3. doi:10.1073/pnas.68.4.820. PMID 5279523. Bibcode: 1971PNAS...68..820K. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=389051
- "Tumor Suppressor (TS) Genes and the Two-Hit Hypothesis | Learn Science at Scitable" (in en). https://www.nature.com/scitable/topicpage/tumor-suppressor-ts-genes-and-the-two-887/.
- "Suppression of human colorectal carcinoma cell growth by wild-type p53". Science 249 (4971): 912–5. 1990. doi:10.1126/science.2144057. PMID 2144057. Bibcode: 1990Sci...249..912B. https://dx.doi.org/10.1126%2Fscience.2144057
- "The murine gene p27Kip1 is haplo-insufficient for tumour suppression". Nature 396 (6707): 177–80. 1998. doi:10.1038/24179. PMID 9823898. Bibcode: 1998Natur.396..177F. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5395202
- Sherr CJ (January 2004). "Principles of tumor suppression". Cell 116 (2): 235–46. doi:10.1016/S0092-8674(03)01075-4. PMID 14744434. https://dx.doi.org/10.1016%2FS0092-8674%2803%2901075-4
- "Metastasis-suppressor genes: a review and perspective on an emerging field". J. Natl. Cancer Inst. 92 (21): 1717–30. November 2000. doi:10.1093/jnci/92.21.1717. PMID 11058615. https://dx.doi.org/10.1093%2Fjnci%2F92.21.1717
- "Cell adhesion system and human cancer morphogenesis". Cancer Sci 94 (7): 575–81. 2003. doi:10.1111/j.1349-7006.2003.tb01485.x. PMID 12841864. https://dx.doi.org/10.1111%2Fj.1349-7006.2003.tb01485.x
- Markowitz S (November 2000). "DNA repair defects inactivate tumor suppressor genes and induce hereditary and sporadic colon cancers". J. Clin. Oncol. 18 (21 Suppl): 75S–80S. PMID 11060332. http://www.ncbi.nlm.nih.gov/pubmed/11060332
- "RETINOBLASTOMA: Protein". http://dpuadweb.depauw.edu/cfornari_web/DISGEN/retinoblastoma_website/public_html/protein.htm.
- Harris, Curtis C. (October 16, 1996). "Structure and Function of p53 Tumor Suppressor Gene: Clues for Rational Cancer Therapeutic Strategies". Journal of the National Cancer Institute 88 (20): 1442–1455. doi:10.1093/jnci/88.20.1442. PMID 8841019. https://dx.doi.org/10.1093%2Fjnci%2F88.20.1442
- "BCL2 (B-Cell Leukemia/Lymphoma 2)". http://atlasgeneticsoncology.org/Genes/GC_BCL2.html.
- Goodsell, David S. (2004-04-01). "The Molecular Perspective: Cytochrome c and Apoptosis" (in en). The Oncologist 9 (2): 226–227. doi:10.1634/theoncologist.9-2-226. ISSN 1083-7159. PMID 15047927. https://dx.doi.org/10.1634%2Ftheoncologist.9-2-226
- Shain, AH; Pollack, JR (2013). "The spectrum of SWI/SNF mutations, ubiquitous in human cancers.". PLOS ONE 8 (1): e55119. doi:10.1371/journal.pone.0055119. PMID 23355908. Bibcode: 2013PLoSO...855119S. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3552954
- "TUMOUR SUPPRESSOR GENES IN CANCER". https://www.letstalkacademy.com/publication/read/tumour-suppressor-genes-in-cancer.