Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
E2F4 was initially described as a transcription factor with a key function in the regulation of cell quiescence. The regulation of E2F4 is complex, as it can be chemically modified through acetylation, from which we present evidence in the brain, as well as methylation, and phosphorylation. The phosphorylation of E2F4 within a conserved threonine motif induces cell cycle re-entry in neurons, while a dominant negative form of E2F4 (E2F4DN), in which the conserved threonines have been substituted by alanines, has been shown to act as a multifactorial therapeutic agent for Alzheimer’s disease (AD).
- acetylated E2F4
- synapsis
- tissue homeostasis
- Alzheimer’s disease
- 5xFAD mice
1. Introduction
E2 factor 4 (E2F4) is a member of the E2F family of transcription factors [1], which are primarily known to regulate the cell cycle. E2F4 was first described as a cell cycle repressor able to interact with p107 [2,3] and p130 [4], two members of the retinoblastoma (RB) family. Nevertheless, its capacity to repress cell cycle progression can be modulated, as it can also facilitate the cell cycle progression of cardiomyocytes, normal intestinal crypt cells, and colorectal cancer cells [5,6]. A number of reviews have been published describing the role of this transcription factor in quiescence and other cell cycle-related mechanisms [7,8,9], and we refer to the reader to these informative reviews for this aspect of E2F4 function.
Interestingly, E2F4 can also play other important roles in cellular physiology, including cell and tissue homeostasis and tissue regeneration [7,8,10,11,12]. Therefore, E2F4 can be considered a multifactorial factor with an important impact on neuronal welfare and brain homeostasis [11,12], suggesting that it may be a promising candidate target for neurodegenerative diseases and brain aging.
E2F4 is a phosphoprotein whose phosphorylation within an evolutionary-conserved threonine motif containing T248 (Figure 1) can modify its function [11,13]. This covalent modification has been targeted by substituting T248 and T250 with alanines, thus resulting in a dominant negative form of E2F4 (E2F4DN). This mutant form, or E2F4, prevents cell cycle re-entry in developing neurons [13] and is able to prevent Alzheimer’s disease (AD)-deleterious processes in 5xFAD mice [11], a murine model of this disease [14].
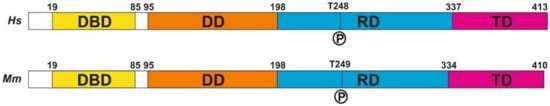
Figure 1. E2F4 structure. The structure of human (Hs) and mouse (Mm) E2F4, derived from NCBI Reference Sequences NP_001941.2 (H. sapiens) and NP_683754.1 (M. musculus). DBD: DNA binding domain, DD: dimerization domain, RD: regulatory domain, TD: transactivation domain.
2. Transcriptional and Non-Transcriptional Functions of E2F4
2.1. E2F4 as a Transcriptional Regulator
Human E2F4 contains 413 amino acids (410 in mouse) distributed throughout four domains (Figure 1). As with other E2F members, it forms a heterodimer with dimerization partner (DP) proteins through its dimerization domain (DD), located at the N-terminus of the molecule. The DD domain is required for its interaction with DNA through the DNA-binding domain (DBD). A third domain located at the C-terminus is required for the function of E2F4 as a transcription factor [16]; this transactivation domain (TD) is blocked when the retinoblastoma (RB) family proteins p107 or p130 interact with E2F4 through its protein-binding domain [10]. This interaction is crucial for the control of the E2F4 function as a transcription factor. Finally, E2F4 has a region that has been proposed as a regulatory domain (RD) [13] in which phosphorylatable residues, such as T248 (see below), are placed. In addition, two nuclear export signals (NES) are present in E2F4, one located within the DBD and the other in the DD [17,18]. These sequences maintain E2F4 within the cytoplasm unless it interacts with p107 or p130, which are required for the translocation of E2F4 to the nucleus. In addition, other factors can induce the translocation of E2F4 to the nucleus, as the latter can also regulate transcription through RB-independent mechanisms [19]. In this regard, E2F4 can interact with KPNB1, RanGAP1, and RanBP2 [19], three proteins that are involved in nuclear import [20,21], and may facilitate E2F4 nuclear translocation in the absence of RB family members. Moreover, E2F4 may be translocated to the nucleus with the help of DP family members DP-2 and DP-3 [22,23,24], likely due to the presence of a nuclear localization signal (NLS) in their sequence, as has been shown in DP-2 [25]. Finally, E2F4 harbors a weak putative NLS in amino acids 52-61 [25], suggesting that E2F4 can translocate into the nucleus in a cofactor-independent manner, similar to E2F5 during keratinocyte differentiation [26].
A ChIP-seq analysis performed in human lymphoblastoid cells identified around 16,000 E2F4 binding sites that potentially regulate 7346 target genes with wide-ranging functions, including cell cycle regulation, DNA repair, RNA processing, stress response, apoptosis, ubiquitination, protein transport and targeting, protein folding, and I-κB kinase/NF-κB cascade regulation [27]. In these cells, E2F4 can bind near transcription start sites (TSSs), a finding confirmed by others [28]. In addition, functional distal sites for E2F4 can be located more than 20 kb away from the annotated TSSs. In both cases, E2F4 can act as an activator as well as a repressor [27]. This analysis also indicated that E2F4 can bind to the promoters of 780 transcription factors, suggesting that E2F4 can indirectly regulate broad classes of genes [27]. Other authors have confirmed that E2F4 can bind to genes related to DNA repair, DNA damage, and G2/M checkpoints, as well as to other classical functions, such as cell cycle regulation, DNA replication, chromosome transactions, and mitotic regulation [29]. In most cases, E2F4 can bind to a specific promoter together with other members of the E2F family [28], indicating that the E2F4 function is subjected to complex cross-regulatory networks [30,31]. Many E2F4 binding sites have been analyzed in specific gene regulatory regions [32]. For instance, the release of a p130-E2F4 complex from sequences immediately upstream of the transcription initiation site of the human CDC2 promoter has been shown to coincide with the induction of CDC2 expression [33].
Several lines of evidence indicate that E2F4 is able to control complex transcriptional regulatory networks in specific cells, thus supporting its multifactorial capacity as a transcription factor. For instance, a combined analysis using gene ontology and expression data has been used to define the network controlled by E2F4 in B cells [34]. In addition, loss-of-function studies on E2F4 silencing using a specific shRNA in acute myeloid leukemia cells have revealed that 276 genes show altered expression patterns in these cells [35]. These E2F4-regulated genes are mostly involved in the regulation of the mitogen-activated protein kinase (MAPK) signaling pathway.
The regulation of gene transcription by E2F4 seems to be mediated through histone acetylation, as E2F4 may interact with CREB binding protein (histone acetyltransferase) [19], and sites where E2F4 binds are histone-modified [27].
2.2. E2F4 and Non-Transcriptional Interactors
E2F4 lacks a strong NLS, which suggests that this protein could play a significant role in the cytoplasm [36]. This is, for instance, the case of the regulation of centriole amplification during multiciliogenesis, which is mediated by the interaction of E2F4 with Deup1 and SAS6, two components of the centriole replication machinery [37]. Indeed, cytoplasmic E2F4 forms organizing centers in multiciliated cells [38]. While centrioles are known to undergo one round of duplication per cell cycle in normal proliferating cells, multiciliated cells show a massive assembly of these organelles during G0, a process initiated by Multicilin in combination with E2F4 (or E2F5) and Dp1 [39,40,41].
The capacity of E2F4 to function out of the nucleus is consistent with a study by Hsu and collaborators [19]. These authors identified a number of E2F4 interactors in mouse embryonic stem cells (mESCs) and a retinal pigment epithelium (RPE)-derived cell line of human origin [19]. Several of these interactors are located outside of the cell nucleus since a cellular component (CC) ontology analysis performed by us using the E2F4 interactors described by Hsu and collaborators [19] confirmed that E2F4 may be functional in the cytoplasm of mESCs and both cytoplasm and extracellular vesicles from RPE-derived cells.
3. Regulation of E2F4 Function by Chemical Modifications
Proteins can be posttranslationally modified through covalent processing events that change their properties, either by proteolytic cleavage or by adding a modifying group, such as acetyl, phosphoryl, glycosyl, and methyl, to one or more amino acids [42]. More than 400 different types of posttranslational modifications [43] affect many aspects of protein function. Some of these chemical modifications have been described in E2F4.
As in the case of other regulators of the cell cycle, E2F4 can be ubiquitinated as a mechanism regulating its protein levels [44]. In addition, E2F4 activity could be modulated by protein acetylation, as observed with another member of the E2F family of transcription factors, E2F1 [45]. E2F1 can be acetylated in sites that lie adjacent to the DBD, thus increasing its DNA-binding ability and activation potential, as well as its protein half-life [45]. In the case of E2F4, Hsu and collaborators [19] demonstrated that both human and mouse E2F4 can be significantly acetylated in K37 and K96. These residues are located within the DBD and DD, respectively, thus suggesting that the capacity for DNA binding and DP heterodimerization of E2F4 can be compromised. This may facilitate the cytoplasmic function of E2F4 as a multifactorial protein. These authors also found small levels of acetylation in K20, K28, K44, K73, K82, K101, K177, K197, K230, and K347 from human E2F4 and in K28, K44, K101, K118, K177, K178, and K339 from mouse E2F4. Most of these residues are located within the DBD and DD of E2F4, suggesting that their acetylation can also participate in the regulation of DNA binding and the DP heterodimerization of E2F4.
Using an acetylated K96-specific antibody, we verified that K96 becomes acetylated in some structures of the adult mouse brain in vivo (Figure 2). This form of acetylated E2F4 can be detected in NeuN-positive cells (i.e., neurons) within the hippocampus (dentate gyrus) (Figure 2a), cerebellum (Figure 2b), and NeuN-negative cells located in the rostral migratory stream (RMS) (Figure 2c), likely neural progenitors. Some NeuN-negative cells in the cerebellum also showed acetylated E2F4-specific immunoreactivity (Figure 2b).
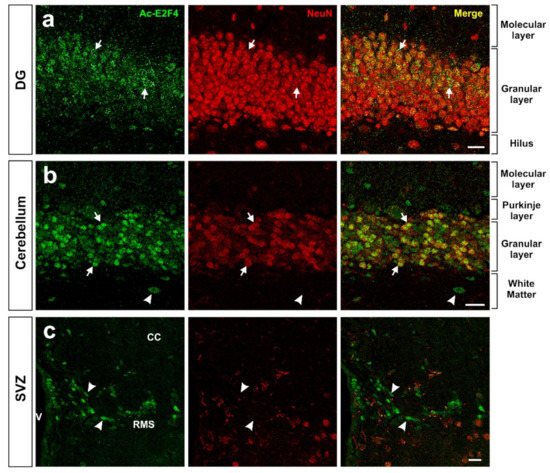
Figure 2. Expression pattern of acetylated E2F4 in the dentate gyrus (DG) (a), cerebellum (b), and subventricular zone (SVZ) (c) of 2.5-month-old WT mice. One single confocal plane showing co-immunostaining with anti-acetylated E2F4 (Ac-E2F4) and anti-NeuN (NeuN) antibodies in sections from the indicated brain areas. NeuN specifically labels neurons. Ac-E2F4 immunostaining in NeuN-positive (arrows) and NeuN-negative (arrowheads) cells is shown. V: ventricle; CC: corpus callosum; RMS: rostral migratory stream. Scale bar: 20 μm.
In non-histone proteins, methylation represents a chemical modification participating in diverse processes, such as cell cycle control, DNA repair, senescence, differentiation, apoptosis, and tumorigenesis [46]. As a multifactorial factor, E2F4 can also become methylated. In this regard, Hsu and collaborators [19] have shown that a significant proportion of K73, K197, and R357 (R360 in mice) residues from E2F4 can be methylated. Interestingly, the methylation of K197 in E2F4 is reminiscent of a similar process in E2F1, affecting K185, which is involved in the regulation of E2F1-induced cell death [46,47,48]. Other residues of human (K20, K37, K53, K57, K74, K96, K101, R147, K177, K230, and K347) and mouse (R297 and K339) E2F4 can also be methylated, as reported by Hsu and collaborators [19].
Finally, the most prominent mechanism regulating E2F4 activity is protein phosphorylation. E2F4 has several residues susceptible to phosphorylation (Figure 1), and several lines of evidence indicate that E2F4 can undergo phosphorylation [49] to modulate its function. In this regard, this chemical modification may regulate E2F4-mediated transcription, either by disrupting its DNA-binding ability, as observed in 3T3 cells [50], or by enhancing the DNA binding of the E2F4/p130 repressor complex, as demonstrated in human fibroblasts [51]. Seven of the theoretical phosphorylation sites of E2F4, including T14, S202, S218, T224, S244, T248, and S384, have been demonstrated to become phosphorylated [52]. Other authors have confirmed the phosphorylation of T14, S218, S244, T248, and S381 in human E2F4 [19], of S218, T224, T249, and S384 in mouse E2F4 [19], and the ortologue of T248/T250 (T261/T263) in chicken E2F4 [13]. In addition, phosphorylation of E2F4 in T249 has been observed in mouse brain extracts using a phosphosite-specific antibody [11], and indirect evidence for the phosphorylation of T248 in the human brain was obtained using a proximity ligation assay with anti-E2F4 and anti-phosphothreonine antibodies [12]. Hsu and collaborators [19] also found evidence of phosphorylation in S16, Y139, S185, S187, S220, S223, and Y389 from human E2F4 and in S75, Y139, T153, S223, S240, T266, Y392, and Y394 from mouse E2F4.
4. E2F4 as a Multifactorial Regulator
4.1. E2F4 as a Regulator of Tissue Homeostasis
In addition to its classical function in regulating quiescence in proliferating cells, E2F4 can also participate in several homeostatic processes. For instance, E2F4 has been associated with the DNA damage checkpoint and repair pathways [29,53,54] (see below), prevention of DNA damage-associated cell death [31], repression of apoptotic genes [55], epigenetics [56], metabolism regulation [57,58], autophagy [59], inflammation [60], and cell repair [61]. In addition, E2F4 function has been associated with oxidative stress [62]. In this regard, the p107-E2F4 complex downregulates PGC-1alpha expression [63], an enzyme that protects cells against oxidative stress and reduces mitochondrial dysfunction in AD [64,65].
The ability of E2F4 to regulate several homeostatic functions may have evolved from its capacity to regulate processes primarily associated with cell cycle arrest and cell differentiation. Indeed, under growth arrest conditions, E2F4 can repress a common set of genes involved in mitochondrial biogenesis and metabolism [66]. Moreover, E2F4 participates in the differentiation of multiple cell types, including the differentiation of myocytes [22,36,67,68,69], neural cells [30,70], adipocytes [71,72,73,74], hematopoietic cells [75], chondrocytes [76], extra-embryonic tissues [77], endothelial cells [78], epithelial cells [79], and multiciliated cells [80,81]. E2F4 can also regulate eye and brain patterning [82,83,84,85], as well as endocytosis and water channel transport in the testes [81].
The capacity of E2F4 to act as a multifactorial factor is likely mediated by the different interactors to which this molecule can bind. In this regard, E2F4 can perform non-canonical actions in cells in the absence of RB family proteins, allowing the transactivation domain to interact with other proteins [19]. After performing biological process (BP) ontology analysis, we found that many E2F4 interactors identified by these authors are related to non-cell cycle processes, including DNA repair, stem cell population maintenance, protein sumoylation in mESCs, as well as retina homeostasis, RNA splicing, organ regeneration, and regulation of lipid kinase activity in RPE-derived cells.
4.2. E2F4 as a Regulator of DNA Repair
Cells have to constantly respond to genotoxic insults that may induce DNA modifications, which usually lead to genome instability. Accumulation of damaged DNA is deleterious for cells since it often results in abnormal proliferation, cell aging, or cell death. Eukaryotic cells have acquired mechanisms of defense against this damage; globally, they are referred to as DNA damage response (DDR), which are in charge of monitoring and removing lesions in their DNA [86]. In this regard, mammalian cells are equipped with several DNA repair pathways, which can be classified into two main groups [87]. On the one hand, the machinery involved in base excision repair, nucleotide excision repair (NER), and mismatch repair can fix single-strand mutations. On the other hand, double strand breaks (DSBs) can be repared through two main mechanisms: homologous recombination (HR), which repairs DSBs during the S-phase or G2 since the sister chromatic is used as a template, and non-homologous end-joining (NHEJ), which is able to repair DSBs at any stage of the cell cycle and in quiescent and postmitotic cells.
DDR can be transcriptionally regulated by E2F factors. These transcription factors usually bind to two adjacent E2F sites within the regulatory regions of genes involved in DNA damage checkpoint and repair [88], thus allowing for functional interactions. Two known E2F factors regulating DDR are E2F4 and E2F1 [27,29], which functionally counteract each other. For instance, E2F4 silencing in MCF7 epithelial breast cells treated with benzoapyrene, an environmental pollutant that triggers DNA damage [89], results in E2F1 derepression and the subsequent induction of DNA repair factors [90]. In primary neurons, the repair response to DSBs is also regulated by E2F1 and E2F4. In this cellular system, E2F1 enhances Cited2 expression, a pro-apoptotic gene required for delayed neuronal cell death, while E2F4 strongly inhibits Cited2 transcription, helping to cell survival [31]. Finally, E2F4 has also been implicated in NER since the p130/E2F4 complex controls the expression of xeroderma pigmentosum complementation group C [53], a protein that serves as the primary initiating factor in the global genome NER pathway [91]. There is also evidence that hypoxia and the anti-angiogenic agent cediranib are both able to induce the binding of p130/E2F4 complexes to E2F consensus sequences in the promoters of homology-directed DNA repair genes, thus reducing gene expression [54,88,92].
In most paradigms, E2F4 seems to act as a repressor of genes involved in DNA damage checkpoint and repair. This function may be favored by the stress kinase p38MAPK, which phosphorylates E2F4 [13] and becomes activated by the DDR [93]. Therefore, the expression of a non-phosphorylatable form of E2F4 (E2F4DN) might modulate the maintenance of the expression of genes involved in DDR.
4.3. E2F4 as a Putative Regulator of Synaptic Function
E2F4 has been related to cognitive impairment [94] and the pathogenesis of AD [95], as well as to other neurological diseases [96], including Parkinson´s disease/mild cognitive impairment [97]. Since AD is largely a synaptic failure [98] occurring prior to cognitive decline or cell death [99], it can be speculated that E2F4 is important for synaptic function.
4.3.1. Transcriptional Regulation of Synaptic Function by E2F4
E2F4 has the potential to regulate the expression of an ample number of synaptic proteins. As evidenced by ChIP-seq datasets from the ENCODE transcription factor targets dataset interrogated with the Harmonizome tool [100], E2F4 can bind to 46 synaptic protein-encoding genes, as well as 127 genes encoding for ion channel subunits. In this regard, there is direct evidence that E2F4 can regulate synaptic function, coming from the transcriptomic analysis performed in mESCs subjected to E2f4 gene knock-out (KO). The transcriptional alterations in synaptic plasticity-related genes upon E2F4 modulation reveal the potential role of this protein in synaptic function. This suggests that E2F4 could be a promising target for several neurological diseases that course with synaptic plasticity impairment, such as AD.
4.3.2. Interaction of E2F4 with Synaptic Regulators
E2F4 can interact with synaptic regulators. We verified using BP ontology that almost half of the E2F4 interactors found in the study by Hsu and collaborators [19], which are common in both mESCs and RPE-derived cells, have a function in either axonal transport or synapse physiology.
The E2F4 interactors involved in synaptic function that were identified in RPE-derived cells include Rac Family Small GTPase 1 (Rac1), cell division cycle 42 (Cdc42), and protein phosphatase 1 catalytic subunit β (PPP1CB) [19]. The actin regulators Rac1 and Cdc42 are important for the structural and functional plasticity of dendritic spines, which are the basis of learning mechanisms [101]. The actin cytoskeleton regulator Rac1 controls synaptic actin dynamics [102] and is involved in actin-regulated short-term presynaptic plasticity through the modulation of synaptic vesicle replenishment [103]. Cdc42 is known to have an important role in dendritic branching [104], and it is part of the mechanism involved in CaMKII activation, which modulates dendritic spine structural plasticity and induces LTP [105]. PPP1CB is one of the three catalytic subunits of protein phosphatase 1 (PP1), a serine/threonine protein phosphatase that regulates synaptic transmission and plasticity [106]. PP1 mediates NMDAR dephosphorylation, modulating the synaptic expression of this receptor [107].
Hsu and collaborators [19] also found Fragile X Mental Retardation Protein (FMRP) to be a candidate cofactor for E2F4 in mESCs. FMRP is an important regulator of activity-dependent plasticity in the brain, and the mutation in the FMR1 gene and subsequent loss of its protein product lead to Fragile X Syndrome (FXS), an inherited cause of autism and intellectual disability [108]. Mechanistically, FMRP is an RNA-binding protein that regulates the synthesis of synaptic and nuclear proteins within various compartments of the neuron [109]. FMRP binds to dendritic mRNA [110], and this may be important in mRNA localization to dendrites [111]. Thus, the hypothetical interaction of E2F4 with FMRP could be responsible for the modulation of synaptic protein transduction.
Hsu and collaborators [19] also found that Snapin, a protein related to synaptic function [112,113], can interact with E2F4 in both mESC and RPE cells.
In addition, the indirect effects of E2F4 on synaptic plasticity have also been described. In this regard, E2F4 can interact with Suv39H1 [114], a histone methyl transferase with an essential role in H3K9me3 methylation that mediates hippocampal memory functions [115].
The interaction of E2F4 with known synaptic regulators suggests that it may modulate synaptic function. This hypothesis is consistent with the observed enrichment of E2F1 in synaptic fractions, which is related to PSD95 expression and becomes upregulated with aging [116]. Furthermore, E2F1 is necessary for de novo neuronal tetraploidization occurring in mice, and this is associated with the alteration of cognition, as mice lacking this transcription factor show enhanced memory acquisition and consolidation [117]. Since E2F1 and E2F4 have antagonistic roles in neuronal function [96], we speculate that E2F4 could facilitate synaptic function and cognition, as opposed to E2F1.
4.3.3. E2F4 and MAPK Proteins in Synaptic Function
Another piece of evidence for the putative capacity of E2F4 to regulate synaptic function comes from the study by [35], which showed that E2F4 can regulate genes involved in the MAPK signaling pathway. Although this pathway has been associated with cancer [35], it is also relevant for synaptic plasticity and AD [118,119,120]. A relevant member of the MAPK family of protein kinases is p38MAPK, the kinase that phosphorylates E2F4 in the Thr conserved motif controlling neuronal tetraploidization [13]. p38MAPK is a protein involved in synaptic plasticity and memory impairment that has been widely related to AD [120,121]. Accordingly, p38MAPK is progressively activated in neurons affected by AD [122] as well as in APP transgenic mice brains [121], and neuronal p38αMAPK mediates synaptic and cognitive dysfunction in a murine model of AD by controlling amyloid-β (Aβ) production [120]. Moreover, downregulation in APP/Tau-transgenic mice of p38MAPK results in the upregulation of genes involved in the MAPK pathway and calcium signaling [121]. Although the implication of E2F4 in this paradigm remains unclear, the expression of some calcium signaling and/or synaptic plasticity-related genes is altered upon p38α-MAPK deficiency in neuronal populations. In particular, the expression of both Grin2a and its encoded protein glutamate ionotropic receptor NMDA type subunit 2A (Grin2a) is decreased, resulting in a reduction of calcium influx in p38α-MAPK-deficient neurons [121]. Finally, knocking down E2f4 using an E2f4-specific shRNA significantly decreased the protein levels of p-ERK [35], a key MAPK that has been involved in both neurodegenerative diseases, as well as in endocannabinoid [123,124,125,126,127,128] and calcium signaling [101,105,129,130,131,132,133], which are critical pathways in synaptic function and modulation.
This entry is adapted from the peer-reviewed paper 10.3390/ijms232012093
This entry is offline, you can click here to edit this entry!