Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Iron is the most abundant transition metal in the human body and a vital micronutrient that is a critical component of many crucial enzymes. Therefore, it is essential to various biological processes, such as DNA synthesis and repair, cell cycle regulation, transport of oxygen, and energy production. Consequently, it is of no surprise that iron levels elevated in cancer and can lead to further tumour development and metastasis
- iron
- iron chelation
- cancer
- drug design
1. Cellular Iron Homeostasis
At physiological pH and oxygen concentration, iron(II) in the body is readily oxidised to iron(III); as a consequence of this, iron is chaperoned to prevent it from partaking in undesirable chemical reactions such as the Haber–Wiess reaction [44]. Once iron has been taken in by the digestive tract, iron is then transported in the plasma by transferrin (Tf). Transferrin binds two ferric ions (Fe3+) and has a high affinity for iron. The iron–transferrin complex binds to transferrin receptor 1 (TfR1) on the surface of a cell that expresses it. The di-ferric transferrin enters the cell by receptor-mediated endocytosis. Once released, iron is reduced by ferrireductase before transportation across the endosomal membrane by the divalent metal transporter1 (DMT1) [45]. Once iron is in the cytoplasm, it is stored in a complex with ferritin (Ft) or used to form Fe-S clusters or heme proteins or contribute to the labile iron pool [46]. Iron can also be transported out of the cell by ferroportin (FPN1). Iron which is neither stored nor transported out of the cell exists in several organelles as the labile iron pool.
The labile iron pool is in a dynamic equilibrium, shifting and changing its constituents depending on the availability of iron, resulting from the influx and efflux of iron from the cell or the stored forms of iron. The iron within this labile iron pool can either be Fe2+ or Fe3+ but is generally accepted to contain more Fe2+ due to the great quantity of water and reductants in cells [47]. Iron within the labile iron pool is bound to intracellular proteins to minimise potentially toxic Fenton chemistry. One of the main intracellular proteins is iron(II)glutathione conjugates [47,48]. The labile iron pool is generally regulated by storing excess iron in a complex with ferritin or exporting it outside the cell with FPN-1. One of the main ways of protecting normal cells from excess labile iron and Fenton chemistry is storing iron in ferritin. For exporting, Fe2+ iron is converted back to Fe3+ by ferroxidase [49]. The molecules Ft and FPN-1 have an iron response element: hairpin loop structures in the 5′ direction on their untranslated region (UTR) of mRNA [50]. These interact with mRNA binding molecules called iron-regulating protein one and iron-regulating protein two (IRP1 and IRP2). When iron conditions are low, IRP1 binds to an iron-responsive element (IRE) in the 5′UTR on the mRNAs of Ft or FPN-1, which blocks, through steric hindrance, the recruitment of ribosomes and obstructs translation [51]. IRP1 binds to the 3′ UTR on TfR1, and DMT1 masks the mRNA to endonuclease digestion, stabilising it. The consequence of the IRP1s actions on iron is to reduce export and increase import, increasing iron levels in the cell. This occurs as IRP1 is an enzyme known as cytosolic aconitase and contains a full [4Fe-4S] cluster in the presence of high iron conditions. However, when intracellular iron levels are low, there is not enough iron for Fe-S biogenesis resulting in a [3Fe-4S] cluster on the enzyme [52]. Therefore, aconitase cannot function enzymatically, resulting in its IRP1 activation. IRP1 therefore utilises a unique iron-sulphur cluster switch to sense iron levels. Simultaneously, low iron conditions result in the ubiquitination of IRP2, resulting in more iron efflux [50]. Through these mechanisms, in normal conditions, the labile iron pool size results from increases or decreases in iron influx and efflux, which are regulated in correspondence to the cells’ needs.
Further to IRP1, iron homeostasis can also be maintained by hepcidin through several mechanisms: it is a hormone that induces FPN-1 degradation, can inhibit recycled iron release from macrophages, absorption in the duodenum and lastly, through iron stored in hepatocytes [53]. Hepcidin expression correlates with both cellular and serum iron levels. When iron levels are high, hepcidin is produced in the liver. This consequently results in the degradation of FPN-1 in duodenal enterocytes and hepatocytes, which prevents these cells from exporting iron [50]. In contrast, this process does not occur when iron levels are low, and the duodenal and hepatocytes can export iron [54].
Mitochondrial Iron Metabolism and Fe-S Biosynthesis
Iron plays a critical role in the mitochondria; however, the exact mechanisms of how the mitochondria receive iron have not been fully elucidated. Mitochondrial iron is the leading destination for cytosolic labile iron. Thus, cytosolic iron from the labile iron pool must cross the outer mitochondrial membrane (OMM) before being utilised for Fe-S cluster synthesis within the mitochondria with its own labile iron pool. The mitochondrial labile iron pool is also susceptible to ROS as the mitochondria are a site of oxygen consumption. There are two hypotheses as to how mitochondrial iron crosses the OMM. The first is that STEAP3 reduces Fe3+ iron in the endosome [55]. The second is the kiss and run theory, where there is the docking of the endosome containing transferrin-bound iron [56]. More evidence is needed to clarify the exact mechanism or mechanisms involved for iron to cross the OMM. However, the mechanisms for iron to travel from the OMM to the inner mitochondrial membrane (IMM) have been elucidated. It involves iron being brought across the IMM by mitoferrin 1 and mitoferrin 2. It has been shown that purified recombinant mitoferrin transports free iron and that its reduced expression results in iron depletion [57,58]. Once transferred across the IMM by mitoferrin 1 and mitoferrin 2, it is incorporated into Fe-S clusters by frataxin and GLRX5 (Glutaredoxin-related protein 5) for many processes, including DNA repair enzymes [59].
The mitochondrial iron sulphur cluster (ISC) assembly machinery starts Fe-S biogenesis. This complex multistep process can be broken down into four main steps. It is crucial not only for the maturation of Fe-S clusters in the mitochondria but also for cytosolic and nuclear Fe-S cluster proteins [60]. The first [2Fe-2S] cluster is synthesised de novo by several vital ISC proteins; this takes place on the ISU1 scaffold protein [61]. Next, an ATP-dependent chaperone protein aids the Fe/S cluster release from Isu1 and its transfer to monothiol glutaredoxin [62]. It can then be used in proteins that require a [2Fe-2S] cluster or passed to the ISC for [4Fe-4S] cluster synthesis; the latter is the third step. Lastly, the fourth step involves the insertion of the [4Fe-4S] cluster into apoproteins that require it by ISC machinery [63]. The labile iron pool has crosstalk between the mitochondrial labile iron pool and the cytosolic, where the demand for Fe-S clusters and heme synthesis in the mitochondria can lower the cytosolic levels of labile iron. The labile iron in both the cytosol and mitochondria that iron chelators target theoretically makes less iron available for incorporation into crucial iron-requiring enzymes.
2. Altered Iron Metabolism in Cancer
Cancer cells have a higher requirement for iron in order to have a proliferation advantage, which is a cancer hallmark [4,5]. Thus, cancers have adapted to increase the overall iron content in the cells. They have achieved this adaption by increasing iron intake and decreasing the efflux, for example, by increasing the transferrin receptor and decreasing ferroportin [64,65,66]. The increase in transferrin can be the result of the oncogene c-Myc. C-Myc is essential as it can be transitionally upregulated in TFR. Alternatively, it can result from hypoxia as iron is needed to deliver oxygen to cells in low oxygen conditions or by IRP2 upregulation [67,68]. Many critical enzymes involved in importing iron are upregulated in cancer, such as membrane receptors such as DMT1, which are overexpressed in several cancers [69]. Proteins that facilitate endosomal uptake of the di ferric transferrin complex are also increased in cancer, and the subsequent STEAP 1–4 proteins that are involved in reducing iron in endosomes are also overexpressed in cancer [46,70,71,72].
As stated above, cancer cells can limit the volume of iron that can efflux; an example of this is the downregulation of ferroportin in several cancers such as ovary, lung myeloma and many more [73,74,75]. Hepcidin can also modulate ferroportin by binding to it, resulting in its eventual degradation; this has shown to be upregulated in prostate cancer cells and shows one method in which ferroportin can be decreased in cancer cells [76]. Irons decreased efflux in cancer cells and increased influx results in a much-increased labile iron pool, as shown in Figure 4. This satisfies its need for iron for the essential process, resulting in further genomic instability by iron-induced ROS. The increased labile iron pools in cancer allow more Fe-S synthesis for crucial cell functions such as DNA repair and replication. Simultaneously, it potentially results in more genetic instability by enhanced Fenton chemistry, aided by the abundance of iron. Therefore, iron chelators have been designed to target labile iron pools, remove iron, and disrupt one of cancer cells’ main adaptive mechanisms.
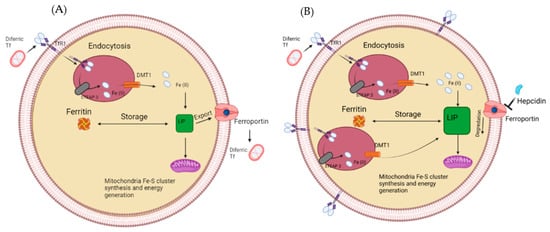
Figure 4. (A) The labile iron pool in a normal healthy cell. Iron enters via the transferrin receptor before getting reduced and used for storage or the labile iron pool (A). (B) How the labile iron pool is altered in cancer cells, upregulated TfR1, STEAP3 and DMT1, while a downregulated Ferroportin results in a larger labile iron pool.
This entry is adapted from the peer-reviewed paper 10.3390/app121910161
This entry is offline, you can click here to edit this entry!