Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Heat shock proteins (HSPs) are conserved molecular chaperones that have cytoprotective properties and are upregulated in response to multiple pathophysiological stresses induced by extensive stimulations, such as high temperature, hypoxia and infectious agents (bacterial and viral). HSPs are categorized into six subfamilies based on the molecular weight: HSP110, HSP90, HSP70, HSP40, small HSPs, as well as chaperonin families.
- heat shock protein
- HSP70s
- HSP90s
- prostate cancer
1. Introduction
Prostate cancer (PCa) is the second most frequently diagnosed cancer in men worldwide, with an incidence of 14.1% and a mortality of 6.8% in 2020 [1]. The incidence rate of PCa has been increasing in the vast majority of countries over recent decades, while death rates have varied greatly worldwide (up to 10-fold) [2]. Despite its high incidence and mortality, the complexity of PCa pathogenesis makes it difficult to offer a radical treatment for PCa patients. Growing evidence has been published to elucidate its pathogenesis associating with androgen/androgen receptor (AR), estrogen, genetic changes, alterations of signal pathways, inflammation and infections, leptin and so on [2]. More mechanistic studies are still necessary.
Heat shock proteins (HSPs) are conserved molecular chaperones that have cytoprotective properties and are upregulated in response to multiple pathophysiological stresses induced by extensive stimulations, such as high temperature, hypoxia and infectious agents (bacterial and viral) [3]. HSPs are categorized into six subfamilies based on the molecular weight: HSP110, HSP90, HSP70, HSP40, small HSPs, as well as chaperonin families [4]. Among all these HSPs, HSP70s (the HSP70 family) and HSP90s (the HSP90 family) are the most two well-studied molecular families with multiple functions. HSP70s are composed of 13 members principally including inducible HSP70 (HSP72 or HSPA1), constitutive heat shock homologous protein 70 (HSC70), glucose-regulated protein 78 (GRP78) and mortalin (GRP75) [5]. HSP90s consist of four members: two in cytosol (HSP90AA1 and HSP90AB1), one in endoplasmic reticulum (ER) (GRP94, also called HSP90B1) and one in mitochondria (TRAP1) [5,6]. The HSP70 and HSP90 family members share the similar regulatory pattern that both require co-chaperones to regulate their functional cycles [7]. These two molecular families are thought to serve as housekeeping genes with basic expression levels under normal conditions; upon exposure to the external stress factors, they are overexpressed to fight against the stressed response. Accumulating evidence has paid attention to the involvement of HSP70 and HSP90 molecular family in multiple biological processes related to tumorigenesis, such as tumor growth, invasion, metastasis. On a cellular level, HSP70s and HSP90s are involved in chaperone activity, protein folding, cell survival and proliferation, cell apoptosis, autophagy, cell differentiation, epithelial–mesenchymal transition (EMT), fibrosis, DNA repair, etc. [8,9,10,11,12] (Figure 1), contributing to the initiation, development and progression of prostate cancer.
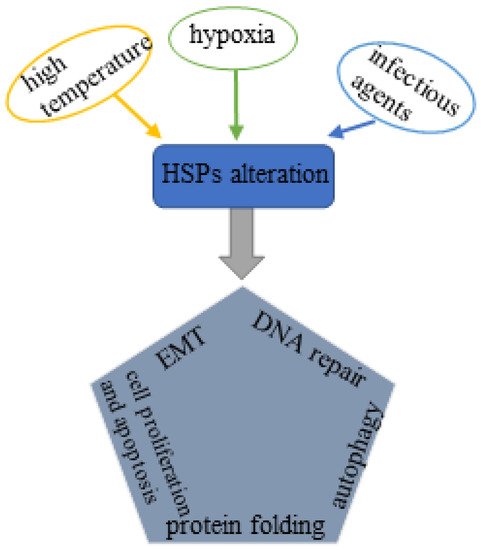
Figure 1. Schematic illustration of the influencing factors of HSPs alteration and its relationship with PCa pathogenesis: External stresses such as hyperthermia, hypoxia and infectious agents stimulate the expression of HSPs, which further affects protein folding, pyroptosis and apoptosis, autophagy, EMT, and DNA repair and ultimately triggers the pathophysiological process of prostate cancer.
2. Prostate Cancer
The malignant transformation of the prostate follows a multi-step process, first prostatic intraepithelial neoplasia (PIN), then localized prostate cancer, followed by locally infiltrated advanced prostate adenocarcinoma, and finally leading to metastatic prostate cancer [13]. The etiology of prostate cancer is complicated and has not been fully elucidated. Known etiologies of prostate cancer include but are not limited to androgen/androgen receptor (AR), EMT, genetics, and pathway alterations.
Androgens regulate the development, maturation, and maintenance of the prostate and affect the proliferation and differentiation of the luminal epithelium. Androgen exposure plays an important role in the occurrence and maintenance of prostate cancer. AR is expressed in almost all primary and metastatic PCa, most of which initially respond to androgen deprivation. Androgen deprivation can improve the prognosis of prostate cancer patients, enabling reduced metastasis and prolonged survival [2]. Thompson and his colleagues treated subjects with finasteride (one of the 5α-reductase inhibitors (5ARI)) and placebo, observing that the incidence rate of prostate cancer was significantly lower in the experimental group than in the control group. 5ARI can reduce the incidence rate of prostate cancer by 24.8% by inhibiting the conversion of testosterone (T) into more effective dihydrotestosterone (DHT) [14].
3. HSP Family
3.1. HSP70 Family
As the most important member in the whole HSP family, HSP70s are upregulated in response to various cellular stresses and able to protect cells from fatal damage [31,32]. HSP70 family molecules have been shown to be involved in cell apoptosis, proliferation, and differentiation. Human HSP70s are widely expressed in a wide range of cell components (such as ER, proteasome, ribosome, mitochondria, and lysosomal membrane). It is believed that the expression of HSP70s is increased in many cancers, such as PCa, breast cancer, which may be associated with the tumorigenesis mechanism [33]. In patients with PCa, serum levels of HSP70s were significantly higher than in patients without PCa [34]. In addition, castration resistant prostate cancer (CRPC) is associated with increased reliance on HSP70s [35], and GRP78 expression is significantly elevated in metastatic CRPC compared with localized prostate cancer [36]. Moreover, GRP75 expression correlates with increased risk of high-grade prostate adenocarcinoma [37]. However, a report published in 2000 indicated that the expression of HSP70 molecules was unaltered in early prostate cancers but was reduced in morphologically advanced cancers compared with non-neoplastic prostate epithelium [38]. These findings suggest that HSP70s may serve as a diagnostic indicator and prognostic indicator for PCa.
4. HSP70 Family in Prostate Cancer Treatment
4.1. Small Molecular Inhibitors
Small molecular inhibitors against different HSP70 family molecules have not been successfully developed for clinical use, with only one agent being subjected to clinical trial. The first well-studied inhibitor, 2-phenylethynesulfonamide (PES) or pifithrin-µ, has been documented to bind to C-terminal SBD of HSP70 and therefore disrupt the association of HSP70s with its co-chaperone HSP40 and other client proteins such as proapoptotic ones APAF-1 and p53 [70]. This interaction of PES with HSP70 molecule induces either autophagic cell death or apoptotic, caspase-dependent, cell death, which depends on different cell types [71,72]. PES is such a potent antitumor agent in vitro and in vivo that this agent shows potent cytotoxicity in multiple cancer cells including LNCaP95 prostate cancer cells, especially when in combination with other inhibitors or medications [73,74,75].
Inhibitors that target N-terminal ATP-binding domain of HSP70s include 15-deoxyspergualin (15-DSG), VER-155008, MKT-077 (the only one subjected to clinical trial) and so on. These agents inhibit HSP70s function by attacking ATP-binding domain of HSP70 and disrupting its ATPase activity, thus blocking cellular proliferation or stimulating cell apoptosis [76,77]. 15-DSG is a natural immunosuppressive agent with a mild effect on cancerous cells compared with other inhibitors [78]. More efficacious agents are MAL3-101, the second-generation inhibitor, and its derivatives [76]. These inhibitors are minimally effective when administered alone for clinical use, but when in combination with other agents, such as HSP90 inhibitor 17-AAG and PS-341(bortezomib), they are shown to have improved efficacy [79]. VER-155008 is an adenosine-derived compound that attacks N-terminal ATP-binding domain of HSP70 family molecules. In vitro studies have demonstrated that VER-155008 is able to induce either caspase-dependent cell death or non-caspase-dependent cell death [77]. The cytotoxic effect of VER-155008 can be improved when administered in combination with HSP90 inhibitors including NVP-AUY922 [80]. The rhodacyanine dye analog MKT-077 is the only agent that has been clinically tested. MKT-077 is thought to serve as a metabolic poison in the mitochondria of cancerous cells where it induces G1 arrest and cell apoptosis [81]. However, the clinical trial of MKT-077 was halted due to the observation of nephrotoxicity, principally renal magnesium wasting. Park and his team recently reported a HSP70 inhibitor apoptosol (AZ) that inhibits the ATPase activity of HSP70s on lysosomal membrane and triggers lysosomal membrane permeabilization (LMP), ultimately inducing lysosome-mediated apoptosis of cancer cells [82,83]. This inhibitor may have higher specificity than above-mentioned HSP70 inhibitors as lysosomal HSP70s are rarely found in normal cells. The derivative of AZ, Az-TPP-O3, was also observed to have potent proapoptotic effect on cancer cells. Az-TPP-O3 primarily attacks the mitochondrial HSP70 molecule mortalin and blocks the interaction of mortalin and p53, which drives a process called mitochondrial outer membrane permeabilization (MOMP) and leads to mitochondria-mediated apoptosis due to caspase activation [82]. Unfortunately, these two unique inhibitors have not been tested in cancer patients, much less in prostate cancer patients. As a result, though all these inhibitors theoretically have strong antitumor activity, temporarily, none of them are appropriate to be developed for clinical use.
4.2. Immunotherapeutic Approaches
A promising strategy to inhibit the expression of HSP70s is the application of monoclonal antibodies (mAb) [79]. The mAb targets HSP70s more accurately with less side-effects than small molecular inhibitors due to its high antigen specificity. A mAb called cmHsp70.1 has completed phase I clinical trial and is being subjected to phase II trial. This monoclonal antibody can recognize the extracellular motif—TKDNNLLGRFELSG (TDK) of membrane HSP70 molecule [84]. In colon cancer, this antibody has been shown to reduce the weight and volume of tumors and promote patients’ survival when administered alone [84].
The development of HSP70 vaccines provides another immunotherapeutic approach for cancers. Several vaccines composed of disease specific epitopes and HSP70 DNA have been successfully developed and subjected to clinical trials. For example, a vaccine called pNGVL4a-Sig/E7(detox)/HSP70 DNA was clinically tested in patients with cervical intraepithelial neoplasia [85]. Another clinical trial has tested the efficacy and toxicity of another vaccine made with HSP70s in chronic myelogenous leukemia [79]. Moreover, a novel immunotherapeutic approach focusing on natural killer (NK) cells based adoptive immunotherapy is being clinically evaluated in which Hsp70-peptide TKD/IL-2 activated, autologous NK cells are employed [79]. To date, no clinical studies of HSP70-related immunotherapy have been performed in patients with prostate cancer. This must be a promising area in the exploration of PCa treatment.
4.3. A Novel Tool with High Specificity: Aptamers
In addition to small molecular inhibitors and antibodies, recent findings have uncovered the inhibitory effect of aptamers, a new category of targeting agents, on functions of HSP70 family molecules. Aptamers (i.e., DNA, RNA aptamers), alongside peptide aptamers, are able to bind to molecules of interest with high specificity and high affinity, exhibiting a functional similarity to antibodies. The differences between two biologics are that aptamers have relatively smaller molecular weight with decreased immunogenicity. Similar to aforesaid small molecular inhibitors, the target site of aptamers is also the NBD or SBD of HSP70 family molecules and the binding of aptamers to the target site can impair the function of HSP70s, as a result of which cancerous cells are susceptible to cell death. Screened from aptamer libraries, A17 has been considered as the most potent aptamer until now. In HeLa cells, A17 is shown to promote cell death (mainly apoptosis) and exhibit its antitumor properties in vivo when combined with cisplatin, but have no such effects when administered alone [86]. A mechanistic study has revealed that this proapoptotic effect of A17 comes from its attack on the NBD of HSP70s that disrupts HSP70 function [86]. Furthermore, the A17 aptamer can inactivate the HSP70 chaperone, but not the one of HSP90 or HSC70 [87].
5. HSP90 Family in Prostate Cancer Treatment
As the most well-studied family of HSPs, HSP90 molecules and their client proteins have been shown to play fundamental roles in tumorigenesis [11]. Targeting HSP90s can lead to the proteasomal degradation of their clients including AR and ultimately influence the signal transduction related to the initiation, development and progression of cancers. This simultaneous inhibitory effects of targeting HSP90 family molecules on AR and other client proteins render HSP90 blockade an attractive therapeutic strategy for prostate cancer [64].
5.1. Natural HSP90 Inhibitors and Their Derivatives
The N-terminal of HSP90 molecules responsible for ATP binding is one of the binding sites for most natural HSP90 inhibitors. These molecular inhibitors ubiquitously have higher affinity for the binding site than natural nucleotides, which are able to inhibit its cycling between ATP- and ADP- bound conformations and impair the functions of HSP90 family molecules [79]. The targeting therapy using HSP90 inhibitors for cancer treatment begins with geldanamycin (GM), an agent derived from Streptomyces hygroscopicus with potent antitumor activity [88]. However, its phase I clinical trial was terminated due to its structural instability and hepatotoxicity. Radicicol (RD), another natural inhibitor derived from Monosporium bonorden, exhibits powerful in vitro antitumor properties by disrupting the critical ATP-binding site of HSP90s, but its structural instability makes it ineffective in vivo. Different from GM and RD, the natural inhibitor Novobiocin destabilizes the client proteins of HSP90 by binding to C-terminal domain of HSP90 and therefore inhibits cancer cell growth [89,90,91]. Importantly, targeting HSP90 C-terminus is one of the approaches to avoid the compensatory heat shock response, induced by targeting the HSP90 ATP-binding site, that stimulates the expression of multiple HSPs, such as HSP90, HSP70, clusterin. Unfortunately, the efficacy and safety of Novobiocin have not been rigorously tested in vivo.
The first generation GM derivatives are 17-AAG (also called tanespimycin or 17-allylamino-17-demethoxygeldanamycin) and 17-DMAG (also called as alvespimycin or 17-dimethylaminoethylamino-17-demethoxygeldanamycin). 17-AAG has been evaluated in the phase II clinical trial, but its poor water solubility and oral bioavailability restricts its development [92]. 17-DMAG shows improved solubility compared with 17-AAG; however, this agent has dose-limiting toxicities: peripheral neuropathy and renal dysfunction [93]. The next generation GM derivative, IPI-504 (retaspimycin), has been shown to have minimal efficacy and unacceptable toxicity in a phase II clinical study in CRPC patients following IPI-504 treatment [94]. This agent was therefore eliminated from therapeutic choices of prostate cancers.
The natural derivatives of RD include NVP-AUY922 and AT13387 which also attack the ATP-binding pocket of HSP90 molecules. NVP-AUY922 is the first generation RD derivative and has strong proapoptotic activity in vitro and powerful antitumor properties in an ex vivo model of prostate cancer. Nevertheless, NVP-AUY922 has not been clinically tested in patients with metastatic CRPC (mCRPC). The second generation derivative, AT13387, shows in vitro and in vivo antitumor activities against prostate cancer, with long duration of action as one of its most important characteristics [64,95]. Theoretically, the use of longer-acting HSP90 inhibitors is more likely to maintain antitumor efficacy with less systemic exposure and side effects [95]. AT13387 has been involved in multiple phase I or II clinical trials, some of which have been completed (NCT01294202, NCT01685268, NCT00878423, NCT01246102) [79]. In addition, a phase I/II clinical trial is in progress to compare the efficacy when administered alone or combined with abiraterone acetate [64]. Recent studies reported a chalcone derivative, SU086, that has potent proapoptotic effect on multiple PCa cell lines including AR-positive cell (C4-2) and AR-negative cell lines (DU145 and PC-3) [96]. This supports a notion that SU086 is active in PCa cells independence of AR status. Mechanistically, SU086 influences the glycolysis process of prostate cancer cells by impairing the function of HSP90 [97]. The clinical value of this inhibitor still needs validation by a number of clinical studies.
5.2. Synthetic HSP90 Inhibitors
Similar to natural inhibitors, the HSP90 N-terminus is also the important target site of synthetic HSP90 inhibitors, among which purine based compounds are one of the choices for drug development [79]. PU-H71 and PU-DZ8 are structurally analogous to ADP but have higher affinity for HSP90. This competitive effect between these two compounds and ADP hinders the binding of ADP and then impairs HSP90 functions. CNF-2024 is also a purine-scaffold compound with high oral bioavailability and has been shown to induce lymphoma cell death [98]. To date, no clinical studies have been performed to evaluate the efficacy and safety of PU series in PCa patients, therefore more clinical studies are warranted.
The screening of ATP-binding proteins using the ATP-affinity column discovered the 2-aminobenzamide derivative SNX-5422 (PF-04929113) [99]. This compound is an orally bioavailable prodrug of PF 04928473 (SNX-2112), a selective HSP90 inhibitor, and has been subjected to phase I clinical trials in patients with solid tumors including PCa as well as hematological malignancies such as lymphomas and chronic lymphocytic leukemia (CLL) [100,101,102]. Due to its ocular toxicity observed in animal models and in a phase I study, the development of SNX-5422 was terminated and waited for further evaluation [102]. Ganetespib (STA-9090), one of the RD derivatives, is a chemically synthetic compound with potent activity against prostate cancer xenografts. A phase II clinical trial, however, shows a negligible efficacy of ganetespib in 17 patients with mCRPC (only 2 patients had progression-free survival (PFS) of more than 4 months) [103]. As a compound targeting HSP90 C-terminus analogous to novobiocin, its analogue KU174 also has powerful antitumor activity against prostate cancer in vitro and in vivo without induction of the heat shock response [104]. Unfortunately, this compound has not been clinically evaluated to date.
This entry is adapted from the peer-reviewed paper 10.3390/life12101489
This entry is offline, you can click here to edit this entry!